 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeExtreme Acceleration of Graph Neural Network-based Prediction Models for Quantum Chemistry
Nov 25, 2022Molecular property calculations are the bedrock of chemical physics. High-fidelity \textit{ab initio} modeling techniques for computing the molecular properties can be prohibitively expensive, and motivate the development of machine-learning models that make the same predictions more efficiently. Training graph neural networks over large molecular databases introduces unique computational challenges such as the need to process millions of small graphs with variable size and support communication patterns that are distinct from learning over large graphs such as social networks. This paper demonstrates a novel hardware-software co-design approach to scale up the training of graph neural networks for molecular property prediction. We introduce an algorithm to coalesce the batches of molecular graphs into fixed size packs to eliminate redundant computation and memory associated with alternative padding techniques and improve throughput via minimizing communication. We demonstrate the effectiveness of our co-design approach by providing an implementation of a well-established molecular property prediction model on the Graphcore Intelligence Processing Units (IPU). We evaluate the training performance on multiple molecular graph databases with varying degrees of graph counts, sizes and sparsity. We demonstrate that such a co-design approach can reduce the training time of such molecular property prediction models from days to less than two hours, opening new possibilities for AI-driven scientific discovery.
Evening the Score: Targeting SARS-CoV-2 Protease Inhibition in Graph Generative Models for Therapeutic Candidates
May 07, 2021
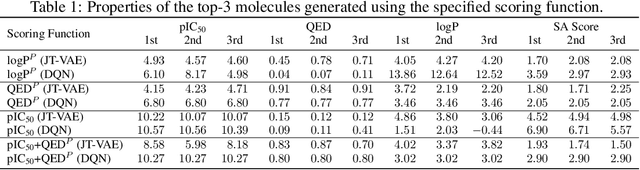

We examine a pair of graph generative models for the therapeutic design of novel drug candidates targeting SARS-CoV-2 viral proteins. Due to a sense of urgency, we chose well-validated models with unique strengths: an autoencoder that generates molecules with similar structures to a dataset of drugs with anti-SARS activity and a reinforcement learning algorithm that generates highly novel molecules. During generation, we explore optimization toward several design targets to balance druglikeness, synthetic accessability, and anti-SARS activity based on \icfifty. This generative framework\footnote{https://github.com/exalearn/covid-drug-design} will accelerate drug discovery in future pandemics through the high-throughput generation of targeted therapeutic candidates.
* arXiv admin note: substantial text overlap with arXiv:2102.04977