 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOptimizing Data Distribution and Kernel Performance for Efficient Training of Chemistry Foundation Models: A Case Study with MACE
Apr 14, 2025



Chemistry Foundation Models (CFMs) that leverage Graph Neural Networks (GNNs) operating on 3D molecular graph structures are becoming indispensable tools for computational chemists and materials scientists. These models facilitate the understanding of matter and the discovery of new molecules and materials. In contrast to GNNs operating on a large homogeneous graphs, GNNs used by CFMs process a large number of geometric graphs of varying sizes, requiring different optimization strategies than those developed for large homogeneous GNNs. This paper presents optimizations for two critical phases of CFM training: data distribution and model training, targeting MACE - a state-of-the-art CFM. We address the challenge of load balancing in data distribution by formulating it as a multi-objective bin packing problem. We propose an iterative algorithm that provides a highly effective, fast, and practical solution, ensuring efficient data distribution. For the training phase, we identify symmetric tensor contraction as the key computational kernel in MACE and optimize this kernel to improve the overall performance. Our combined approach of balanced data distribution and kernel optimization significantly enhances the training process of MACE. Experimental results demonstrate a substantial speedup, reducing per-epoch execution time for training from 12 to 2 minutes on 740 GPUs with a 2.6M sample dataset.
Thinking Fast and Laterally: Multi-Agentic Approach for Reasoning about Uncertain Emerging Events
Dec 10, 2024This paper introduces lateral thinking to implement System-2 reasoning capabilities in AI systems, focusing on anticipatory and causal reasoning under uncertainty. We present a framework for systematic generation and modeling of lateral thinking queries and evaluation datasets. We introduce Streaming Agentic Lateral Thinking (SALT), a multi-agent framework designed to process complex, low-specificity queries in streaming data environments. SALT implements lateral thinking-inspired System-2 reasoning through a dynamic communication structure between specialized agents. Our key insight is that lateral information flow across long-distance agent interactions, combined with fine-grained belief management, yields richer information contexts and enhanced reasoning. Preliminary quantitative and qualitative evaluations indicate SALT's potential to outperform single-agent systems in handling complex lateral reasoning tasks in a streaming environment.
ChemReasoner: Heuristic Search over a Large Language Model's Knowledge Space using Quantum-Chemical Feedback
Feb 21, 2024



The discovery of new catalysts is essential for the design of new and more efficient chemical processes in order to transition to a sustainable future. We introduce an AI-guided computational screening framework unifying linguistic reasoning with quantum-chemistry based feedback from 3D atomistic representations. Our approach formulates catalyst discovery as an uncertain environment where an agent actively searches for highly effective catalysts via the iterative combination of large language model (LLM)-derived hypotheses and atomistic graph neural network (GNN)-derived feedback. Identified catalysts in intermediate search steps undergo structural evaluation based on spatial orientation, reaction pathways, and stability. Scoring functions based on adsorption energies and barriers steer the exploration in the LLM's knowledge space toward energetically favorable, high-efficiency catalysts. We introduce planning methods that automatically guide the exploration without human input, providing competitive performance against expert-enumerated chemical descriptor-based implementations. By integrating language-guided reasoning with computational chemistry feedback, our work pioneers AI-accelerated, trustworthy catalyst discovery.
GLaM: Fine-Tuning Large Language Models for Domain Knowledge Graph Alignment via Neighborhood Partitioning and Generative Subgraph Encoding
Feb 16, 2024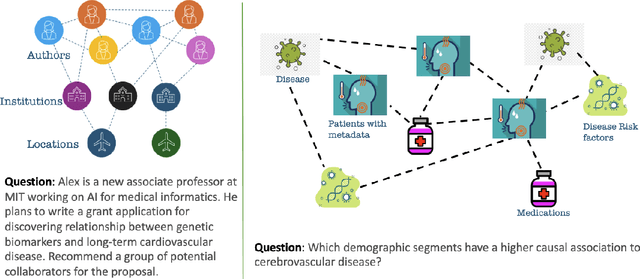
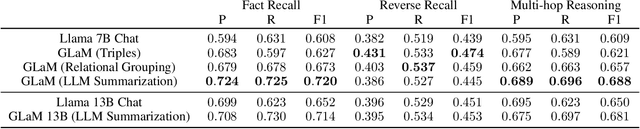
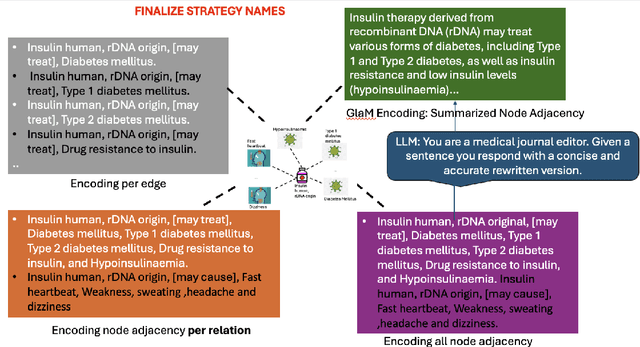
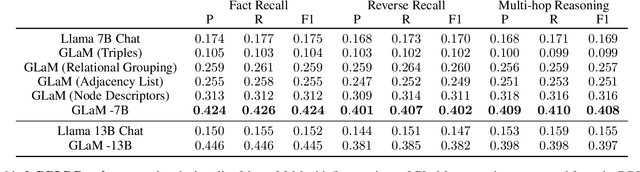
Integrating large language models (LLMs) with knowledge graphs derived from domain-specific data represents an important advancement towards more powerful and factual reasoning. As these models grow more capable, it is crucial to enable them to perform multi-step inferences over real-world knowledge graphs while minimizing hallucination. While large language models excel at conversation and text generation, their ability to reason over domain-specialized graphs of interconnected entities remains limited. For example, can we query a LLM to identify the optimal contact in a professional network for a specific goal, based on relationships and attributes in a private database? The answer is no--such capabilities lie beyond current methods. However, this question underscores a critical technical gap that must be addressed. Many high-value applications in areas such as science, security, and e-commerce rely on proprietary knowledge graphs encoding unique structures, relationships, and logical constraints. We introduce a fine-tuning framework for developing Graph-aligned LAnguage Models (GLaM) that transforms a knowledge graph into an alternate text representation with labeled question-answer pairs. We demonstrate that grounding the models in specific graph-based knowledge expands the models' capacity for structure-based reasoning. Our methodology leverages the large-language model's generative capabilities to create the dataset and proposes an efficient alternate to retrieval-augmented generation styled methods.
Evaluating Emerging AI/ML Accelerators: IPU, RDU, and NVIDIA/AMD GPUs
Nov 08, 2023The relentless advancement of artificial intelligence (AI) and machine learning (ML) applications necessitates the development of specialized hardware accelerators capable of handling the increasing complexity and computational demands. Traditional computing architectures, based on the von Neumann model, are being outstripped by the requirements of contemporary AI/ML algorithms, leading to a surge in the creation of accelerators like the Graphcore Intelligence Processing Unit (IPU), Sambanova Reconfigurable Dataflow Unit (RDU), and enhanced GPU platforms. These hardware accelerators are characterized by their innovative data-flow architectures and other design optimizations that promise to deliver superior performance and energy efficiency for AI/ML tasks. This research provides a preliminary evaluation and comparison of these commercial AI/ML accelerators, delving into their hardware and software design features to discern their strengths and unique capabilities. By conducting a series of benchmark evaluations on common DNN operators and other AI/ML workloads, we aim to illuminate the advantages of data-flow architectures over conventional processor designs and offer insights into the performance trade-offs of each platform. The findings from our study will serve as a valuable reference for the design and performance expectations of research prototypes, thereby facilitating the development of next-generation hardware accelerators tailored for the ever-evolving landscape of AI/ML applications. Through this analysis, we aspire to contribute to the broader understanding of current accelerator technologies and to provide guidance for future innovations in the field.
Monte Carlo Thought Search: Large Language Model Querying for Complex Scientific Reasoning in Catalyst Design
Oct 22, 2023



Discovering novel catalysts requires complex reasoning involving multiple chemical properties and resultant trade-offs, leading to a combinatorial growth in the search space. While large language models (LLM) have demonstrated novel capabilities for chemistry through complex instruction following capabilities and high quality reasoning, a goal-driven combinatorial search using LLMs has not been explored in detail. In this work, we present a Monte Carlo Tree Search-based approach that improves beyond state-of-the-art chain-of-thought prompting variants to augment scientific reasoning. We introduce two new reasoning datasets: 1) a curation of computational chemistry simulations, and 2) diverse questions written by catalysis researchers for reasoning about novel chemical conversion processes. We improve over the best baseline by 25.8\% and find that our approach can augment scientist's reasoning and discovery process with novel insights.
Robust Explanations for Deep Neural Networks via Pseudo Neural Tangent Kernel Surrogate Models
May 23, 2023
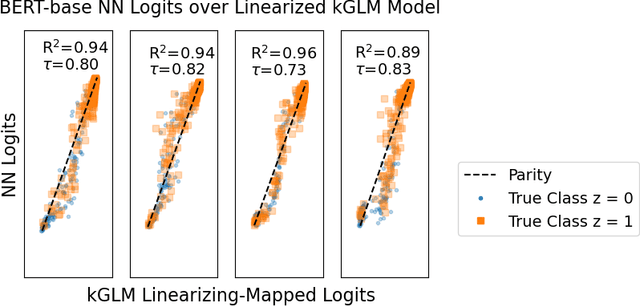
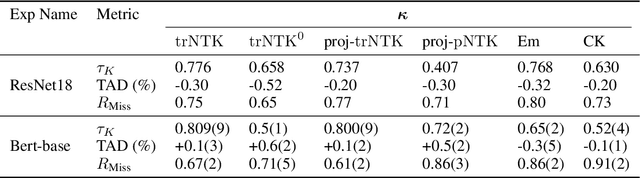
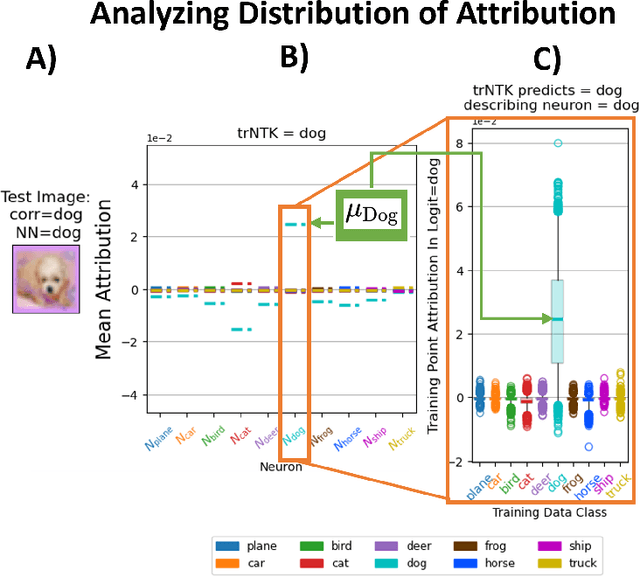
One of the ways recent progress has been made on explainable AI has been via explain-by-example strategies, specifically, through data attribution tasks. The feature spaces used to attribute decisions to training data, however, have not been compared against one another as to whether they form a truly representative surrogate model of the neural network (NN). Here, we demonstrate the efficacy of surrogate linear feature spaces to neural networks through two means: (1) we establish that a normalized psuedo neural tangent kernel (pNTK) is more correlated to the neural network decision functions than embedding based and influence based alternatives in both computer vision and large language model architectures; (2) we show that the attributions created from the normalized pNTK more accurately select perturbed training data in a data poisoning attribution task than these alternatives. Based on these observations, we conclude that kernel linear models are effective surrogate models across multiple classification architectures and that pNTK-based kernels are the most appropriate surrogate feature space of all kernels studied.
BitGNN: Unleashing the Performance Potential of Binary Graph Neural Networks on GPUs
May 04, 2023



Recent studies have shown that Binary Graph Neural Networks (GNNs) are promising for saving computations of GNNs through binarized tensors. Prior work, however, mainly focused on algorithm designs or training techniques, leaving it open to how to materialize the performance potential on accelerator hardware fully. This work redesigns the binary GNN inference backend from the efficiency perspective. It fills the gap by proposing a series of abstractions and techniques to map binary GNNs and their computations best to fit the nature of bit manipulations on GPUs. Results on real-world graphs with GCNs, GraphSAGE, and GraphSAINT show that the proposed techniques outperform state-of-the-art binary GNN implementations by 8-22X with the same accuracy maintained. BitGNN code is publicly available.
Cloud Services Enable Efficient AI-Guided Simulation Workflows across Heterogeneous Resources
Mar 15, 2023Applications that fuse machine learning and simulation can benefit from the use of multiple computing resources, with, for example, simulation codes running on highly parallel supercomputers and AI training and inference tasks on specialized accelerators. Here, we present our experiences deploying two AI-guided simulation workflows across such heterogeneous systems. A unique aspect of our approach is our use of cloud-hosted management services to manage challenging aspects of cross-resource authentication and authorization, function-as-a-service (FaaS) function invocation, and data transfer. We show that these methods can achieve performance parity with systems that rely on direct connection between resources. We achieve parity by integrating the FaaS system and data transfer capabilities with a system that passes data by reference among managers and workers, and a user-configurable steering algorithm to hide data transfer latencies. We anticipate that this ease of use can enable routine use of heterogeneous resources in computational science.
Extreme Acceleration of Graph Neural Network-based Prediction Models for Quantum Chemistry
Nov 25, 2022Molecular property calculations are the bedrock of chemical physics. High-fidelity \textit{ab initio} modeling techniques for computing the molecular properties can be prohibitively expensive, and motivate the development of machine-learning models that make the same predictions more efficiently. Training graph neural networks over large molecular databases introduces unique computational challenges such as the need to process millions of small graphs with variable size and support communication patterns that are distinct from learning over large graphs such as social networks. This paper demonstrates a novel hardware-software co-design approach to scale up the training of graph neural networks for molecular property prediction. We introduce an algorithm to coalesce the batches of molecular graphs into fixed size packs to eliminate redundant computation and memory associated with alternative padding techniques and improve throughput via minimizing communication. We demonstrate the effectiveness of our co-design approach by providing an implementation of a well-established molecular property prediction model on the Graphcore Intelligence Processing Units (IPU). We evaluate the training performance on multiple molecular graph databases with varying degrees of graph counts, sizes and sparsity. We demonstrate that such a co-design approach can reduce the training time of such molecular property prediction models from days to less than two hours, opening new possibilities for AI-driven scientific discovery.