 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOptimizing Data Distribution and Kernel Performance for Efficient Training of Chemistry Foundation Models: A Case Study with MACE
Apr 14, 2025



Chemistry Foundation Models (CFMs) that leverage Graph Neural Networks (GNNs) operating on 3D molecular graph structures are becoming indispensable tools for computational chemists and materials scientists. These models facilitate the understanding of matter and the discovery of new molecules and materials. In contrast to GNNs operating on a large homogeneous graphs, GNNs used by CFMs process a large number of geometric graphs of varying sizes, requiring different optimization strategies than those developed for large homogeneous GNNs. This paper presents optimizations for two critical phases of CFM training: data distribution and model training, targeting MACE - a state-of-the-art CFM. We address the challenge of load balancing in data distribution by formulating it as a multi-objective bin packing problem. We propose an iterative algorithm that provides a highly effective, fast, and practical solution, ensuring efficient data distribution. For the training phase, we identify symmetric tensor contraction as the key computational kernel in MACE and optimize this kernel to improve the overall performance. Our combined approach of balanced data distribution and kernel optimization significantly enhances the training process of MACE. Experimental results demonstrate a substantial speedup, reducing per-epoch execution time for training from 12 to 2 minutes on 740 GPUs with a 2.6M sample dataset.
Revealing the Evolution of Order in Materials Microstructures Using Multi-Modal Computer Vision
Nov 15, 2024



The development of high-performance materials for microelectronics, energy storage, and extreme environments depends on our ability to describe and direct property-defining microstructural order. Our present understanding is typically derived from laborious manual analysis of imaging and spectroscopy data, which is difficult to scale, challenging to reproduce, and lacks the ability to reveal latent associations needed for mechanistic models. Here, we demonstrate a multi-modal machine learning (ML) approach to describe order from electron microscopy analysis of the complex oxide La$_{1-x}$Sr$_x$FeO$_3$. We construct a hybrid pipeline based on fully and semi-supervised classification, allowing us to evaluate both the characteristics of each data modality and the value each modality adds to the ensemble. We observe distinct differences in the performance of uni- and multi-modal models, from which we draw general lessons in describing crystal order using computer vision.
Reducing Down(stream)time: Pretraining Molecular GNNs using Heterogeneous AI Accelerators
Nov 08, 2022The demonstrated success of transfer learning has popularized approaches that involve pretraining models from massive data sources and subsequent finetuning towards a specific task. While such approaches have become the norm in fields such as natural language processing, implementation and evaluation of transfer learning approaches for chemistry are in the early stages. In this work, we demonstrate finetuning for downstream tasks on a graph neural network (GNN) trained over a molecular database containing 2.7 million water clusters. The use of Graphcore IPUs as an AI accelerator for training molecular GNNs reduces training time from a reported 2.7 days on 0.5M clusters to 1.2 hours on 2.7M clusters. Finetuning the pretrained model for downstream tasks of molecular dynamics and transfer to a different potential energy surface took only 8.3 hours and 28 minutes, respectively, on a single GPU.
Decoding the Protein-ligand Interactions Using Parallel Graph Neural Networks
Nov 30, 2021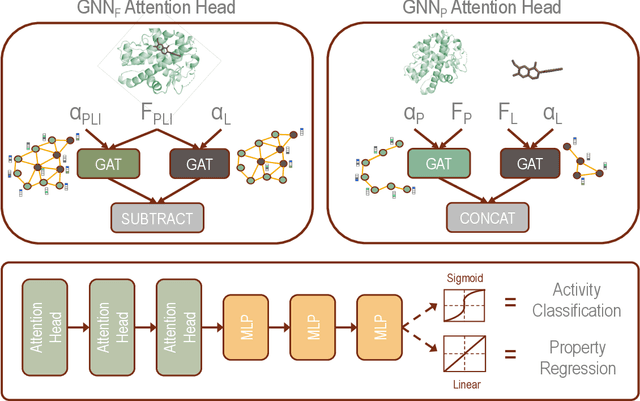
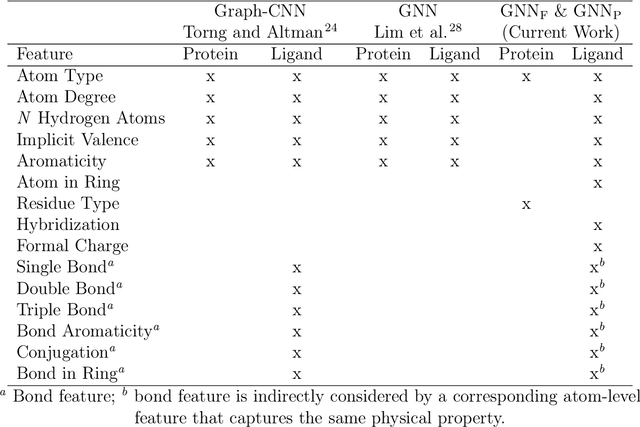

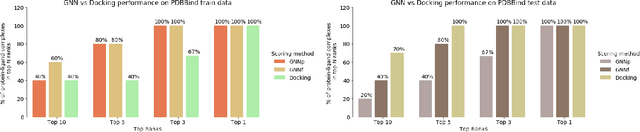
Protein-ligand interactions (PLIs) are fundamental to biochemical research and their identification is crucial for estimating biophysical and biochemical properties for rational therapeutic design. Currently, experimental characterization of these properties is the most accurate method, however, this is very time-consuming and labor-intensive. A number of computational methods have been developed in this context but most of the existing PLI prediction heavily depends on 2D protein sequence data. Here, we present a novel parallel graph neural network (GNN) to integrate knowledge representation and reasoning for PLI prediction to perform deep learning guided by expert knowledge and informed by 3D structural data. We develop two distinct GNN architectures, GNNF is the base implementation that employs distinct featurization to enhance domain-awareness, while GNNP is a novel implementation that can predict with no prior knowledge of the intermolecular interactions. The comprehensive evaluation demonstrated that GNN can successfully capture the binary interactions between ligand and proteins 3D structure with 0.979 test accuracy for GNNF and 0.958 for GNNP for predicting activity of a protein-ligand complex. These models are further adapted for regression tasks to predict experimental binding affinities and pIC50 is crucial for drugs potency and efficacy. We achieve a Pearson correlation coefficient of 0.66 and 0.65 on experimental affinity and 0.50 and 0.51 on pIC50 with GNNF and GNNP, respectively, outperforming similar 2D sequence-based models. Our method can serve as an interpretable and explainable artificial intelligence (AI) tool for predicted activity, potency, and biophysical properties of lead candidates. To this end, we show the utility of GNNP on SARS-Cov-2 protein targets by screening a large compound library and comparing our prediction with the experimentally measured data.
Geometric learning of the conformational dynamics of molecules using dynamic graph neural networks
Jun 24, 2021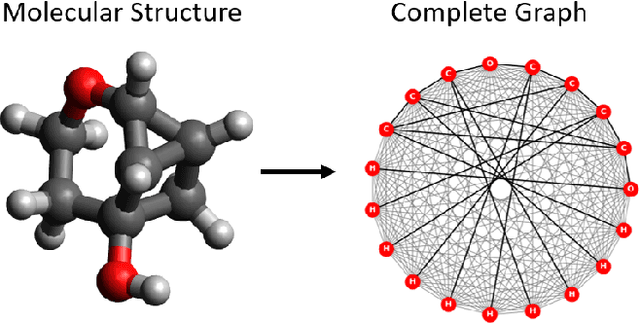
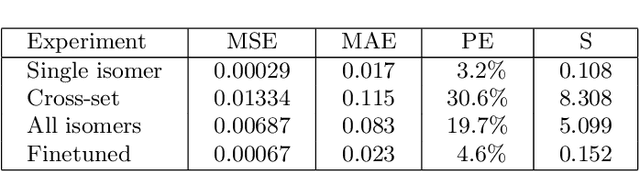
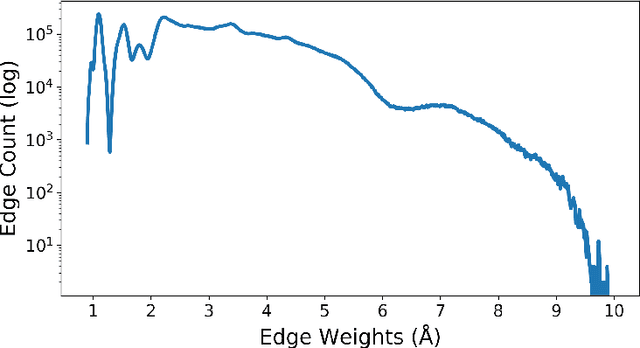
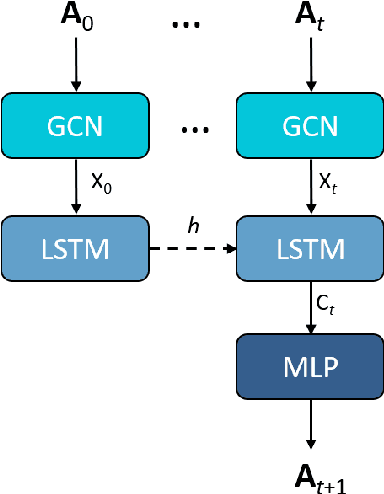
We apply a temporal edge prediction model for weighted dynamic graphs to predict time-dependent changes in molecular structure. Each molecule is represented as a complete graph in which each atom is a vertex and all vertex pairs are connected by an edge weighted by the Euclidean distance between atom pairs. We ingest a sequence of complete molecular graphs into a dynamic graph neural network (GNN) to predict the graph at the next time step. Our dynamic GNN predicts atom-to-atom distances with a mean absolute error of 0.017 \r{A}, which is considered ``chemically accurate'' for molecular simulations. We also explored the transferability of a trained network to new molecular systems and found that finetuning with less than 10% of the total trajectory provides a mean absolute error of the same order of magnitude as that when training from scratch on the full molecular trajectory.
Benchmarking Deep Graph Generative Models for Optimizing New Drug Molecules for COVID-19
Feb 09, 2021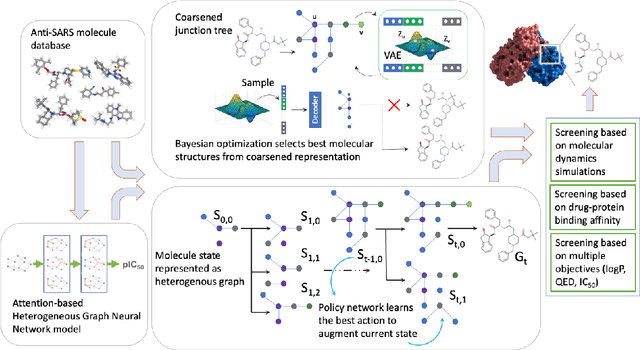
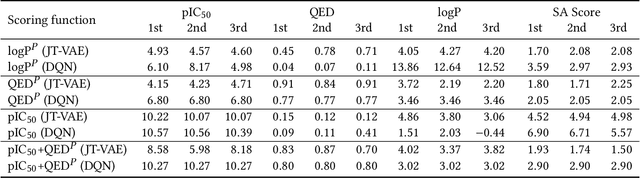
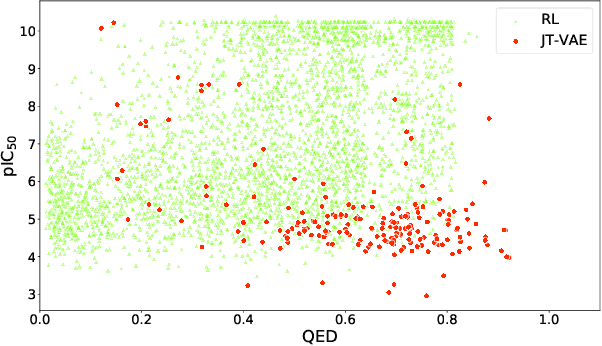
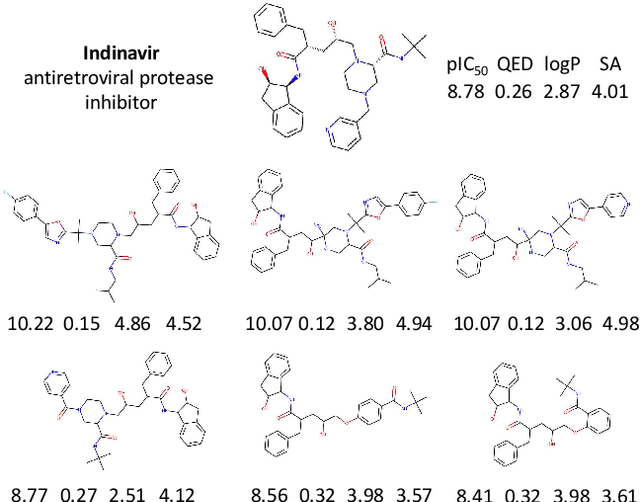
Design of new drug compounds with target properties is a key area of research in generative modeling. We present a small drug molecule design pipeline based on graph-generative models and a comparison study of two state-of-the-art graph generative models for designing COVID-19 targeted drug candidates: 1) a variational autoencoder-based approach (VAE) that uses prior knowledge of molecules that have been shown to be effective for earlier coronavirus treatments and 2) a deep Q-learning method (DQN) that generates optimized molecules without any proximity constraints. We evaluate the novelty of the automated molecule generation approaches by validating the candidate molecules with drug-protein binding affinity models. The VAE method produced two novel molecules with similar structures to the antiretroviral protease inhibitor Indinavir that show potential binding affinity for the SARS-CoV-2 protein target 3-chymotrypsin-like protease (3CL-protease).
HydroNet: Benchmark Tasks for Preserving Intermolecular Interactions and Structural Motifs in Predictive and Generative Models for Molecular Data
Nov 30, 2020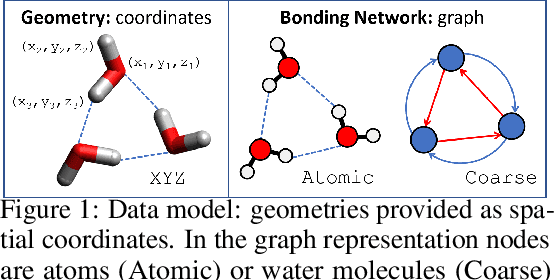
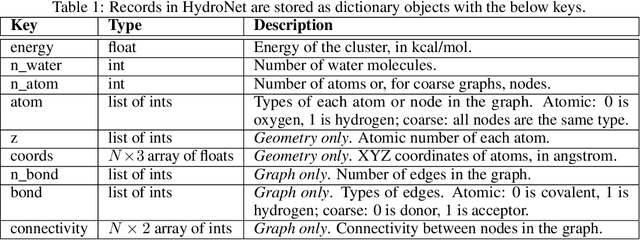
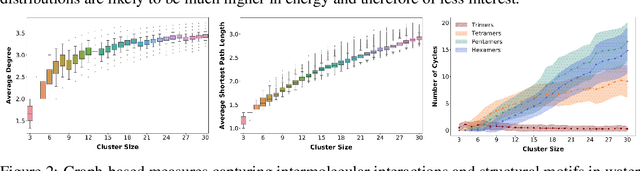
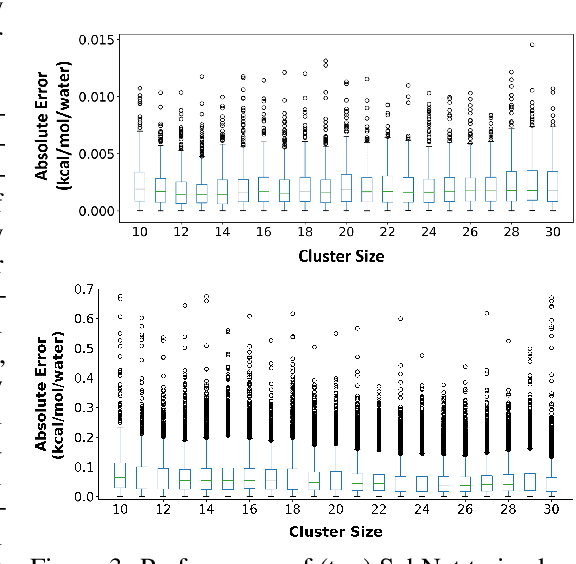
Intermolecular and long-range interactions are central to phenomena as diverse as gene regulation, topological states of quantum materials, electrolyte transport in batteries, and the universal solvation properties of water. We present a set of challenge problems for preserving intermolecular interactions and structural motifs in machine-learning approaches to chemical problems, through the use of a recently published dataset of 4.95 million water clusters held together by hydrogen bonding interactions and resulting in longer range structural patterns. The dataset provides spatial coordinates as well as two types of graph representations, to accommodate a variety of machine-learning practices.