 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDynamic Molecular Graph-based Implementation for Biophysical Properties Prediction
Dec 20, 2022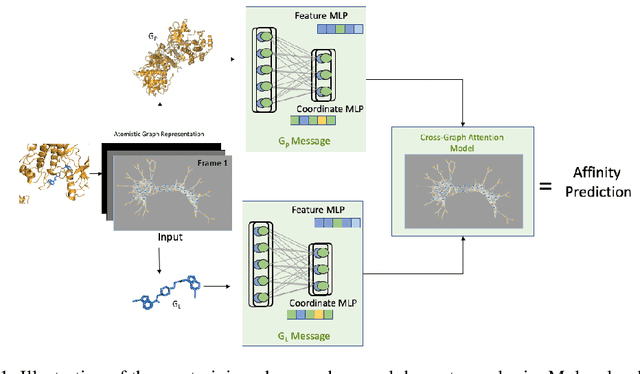
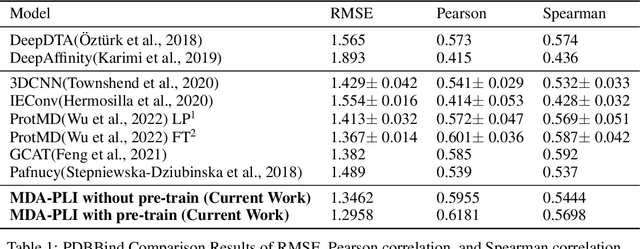
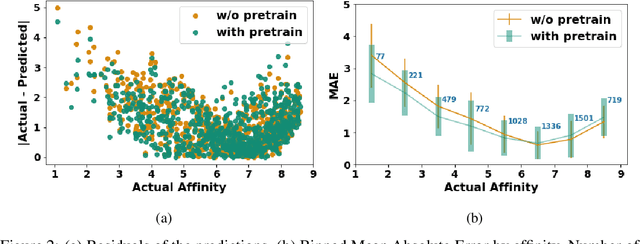
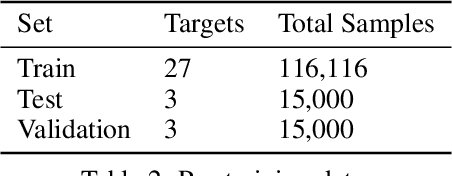
Neural Networks (GNNs) have revolutionized the molecular discovery to understand patterns and identify unknown features that can aid in predicting biophysical properties and protein-ligand interactions. However, current models typically rely on 2-dimensional molecular representations as input, and while utilization of 2\3- dimensional structural data has gained deserved traction in recent years as many of these models are still limited to static graph representations. We propose a novel approach based on the transformer model utilizing GNNs for characterizing dynamic features of protein-ligand interactions. Our message passing transformer pre-trains on a set of molecular dynamic data based off of physics-based simulations to learn coordinate construction and make binding probability and affinity predictions as a downstream task. Through extensive testing we compare our results with the existing models, our MDA-PLI model was able to outperform the molecular interaction prediction models with an RMSE of 1.2958. The geometric encodings enabled by our transformer architecture and the addition of time series data add a new dimensionality to this form of research.
Decoding the Protein-ligand Interactions Using Parallel Graph Neural Networks
Nov 30, 2021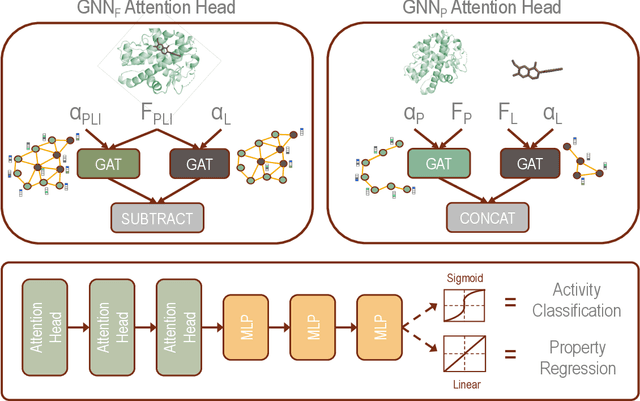
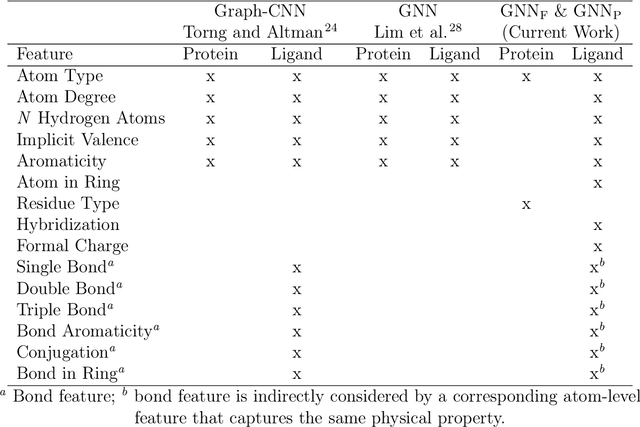

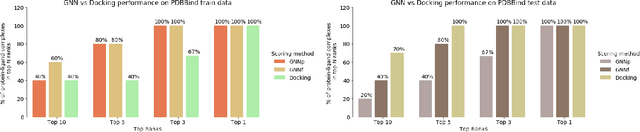
Protein-ligand interactions (PLIs) are fundamental to biochemical research and their identification is crucial for estimating biophysical and biochemical properties for rational therapeutic design. Currently, experimental characterization of these properties is the most accurate method, however, this is very time-consuming and labor-intensive. A number of computational methods have been developed in this context but most of the existing PLI prediction heavily depends on 2D protein sequence data. Here, we present a novel parallel graph neural network (GNN) to integrate knowledge representation and reasoning for PLI prediction to perform deep learning guided by expert knowledge and informed by 3D structural data. We develop two distinct GNN architectures, GNNF is the base implementation that employs distinct featurization to enhance domain-awareness, while GNNP is a novel implementation that can predict with no prior knowledge of the intermolecular interactions. The comprehensive evaluation demonstrated that GNN can successfully capture the binary interactions between ligand and proteins 3D structure with 0.979 test accuracy for GNNF and 0.958 for GNNP for predicting activity of a protein-ligand complex. These models are further adapted for regression tasks to predict experimental binding affinities and pIC50 is crucial for drugs potency and efficacy. We achieve a Pearson correlation coefficient of 0.66 and 0.65 on experimental affinity and 0.50 and 0.51 on pIC50 with GNNF and GNNP, respectively, outperforming similar 2D sequence-based models. Our method can serve as an interpretable and explainable artificial intelligence (AI) tool for predicted activity, potency, and biophysical properties of lead candidates. To this end, we show the utility of GNNP on SARS-Cov-2 protein targets by screening a large compound library and comparing our prediction with the experimentally measured data.