 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDC-W2S: Dual-Consensus Weak-to-Strong Training for Reliable Process Reward Modeling in Biological Reasoning
Mar 09, 2026In scientific reasoning tasks, the veracity of the reasoning process is as critical as the final outcome. While Process Reward Models (PRMs) offer a solution to the coarse-grained supervision problems inherent in Outcome Reward Models (ORMs), their deployment is hindered by the prohibitive cost of obtaining expert-verified step-wise labels. This paper addresses the challenge of training reliable PRMs using abundant but noisy "weak" supervision. We argue that existing Weak-to-Strong Generalization (W2SG) theories lack prescriptive guidelines for selecting high-quality training signals from noisy data. To bridge this gap, we introduce the Dual-Consensus Weak-to-Strong (DC-W2S) framework. By intersecting Self-Consensus (SC) metrics among weak supervisors with Neighborhood-Consensus (NC) metrics in the embedding space, we stratify supervision signals into distinct reliability regimes. We then employ a curriculum of instance-level balanced sampling and label-level reliability-aware masking to guide the training process. We demonstrate that DC-W2S enables the training of robust PRMs for complex reasoning without exhaustive expert annotation, proving that strategic data curation is more effective than indiscriminate training on large-scale noisy datasets.
oMeBench: Towards Robust Benchmarking of LLMs in Organic Mechanism Elucidation and Reasoning
Oct 09, 2025Organic reaction mechanisms are the stepwise elementary reactions by which reactants form intermediates and products, and are fundamental to understanding chemical reactivity and designing new molecules and reactions. Although large language models (LLMs) have shown promise in understanding chemical tasks such as synthesis design, it is unclear to what extent this reflects genuine chemical reasoning capabilities, i.e., the ability to generate valid intermediates, maintain chemical consistency, and follow logically coherent multi-step pathways. We address this by introducing oMeBench, the first large-scale, expert-curated benchmark for organic mechanism reasoning in organic chemistry. It comprises over 10,000 annotated mechanistic steps with intermediates, type labels, and difficulty ratings. Furthermore, to evaluate LLM capability more precisely and enable fine-grained scoring, we propose oMeS, a dynamic evaluation framework that combines step-level logic and chemical similarity. We analyze the performance of state-of-the-art LLMs, and our results show that although current models display promising chemical intuition, they struggle with correct and consistent multi-step reasoning. Notably, we find that using prompting strategy and fine-tuning a specialist model on our proposed dataset increases performance by 50% over the leading closed-source model. We hope that oMeBench will serve as a rigorous foundation for advancing AI systems toward genuine chemical reasoning.
mCLM: A Function-Infused and Synthesis-Friendly Modular Chemical Language Model
May 18, 2025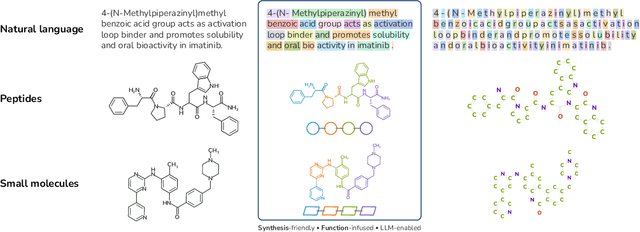
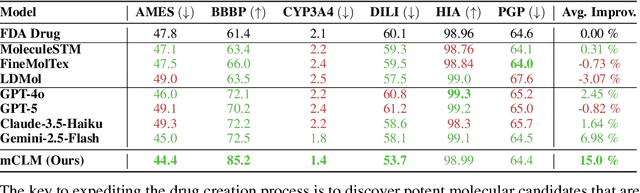
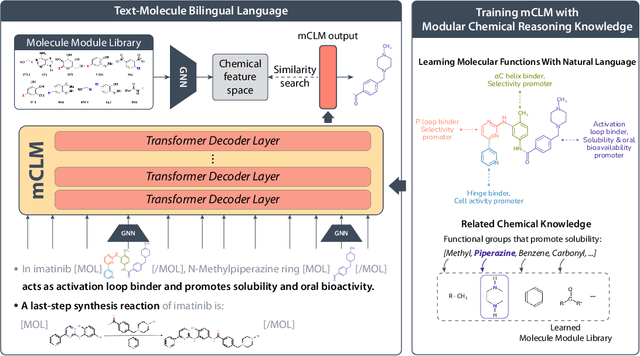
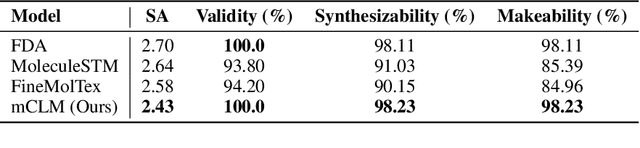
Despite their ability to understand chemical knowledge and accurately generate sequential representations, large language models (LLMs) remain limited in their capacity to propose novel molecules with drug-like properties. In addition, the molecules that LLMs propose can often be challenging to make in the lab. To more effectively enable the discovery of functional small molecules, LLMs need to learn a molecular language. However, LLMs are currently limited by encoding molecules from atoms. In this paper, we argue that just like tokenizing texts into (sub-)word tokens instead of characters, molecules should be decomposed and reassembled at the level of functional building blocks, i.e., parts of molecules that bring unique functions and serve as effective building blocks for real-world automated laboratory synthesis. This motivates us to propose mCLM, a modular Chemical-Language Model tokenizing molecules into building blocks and learning a bilingual language model of both natural language descriptions of functions and molecule building blocks. By reasoning on such functional building blocks, mCLM guarantees to generate efficiently synthesizable molecules thanks to recent progress in block-based chemistry, while also improving the functions of molecules in a principled manner. In experiments on 430 FDA-approved drugs, we find mCLM capable of significantly improving 5 out of 6 chemical functions critical to determining drug potentials. More importantly, mCLM can reason on multiple functions and improve the FDA-rejected drugs (``fallen angels'') over multiple iterations to greatly improve their shortcomings.
Class-based Subset Selection for Transfer Learning under Extreme Label Shift
Dec 30, 2024



Existing work within transfer learning often follows a two-step process -- pre-training over a large-scale source domain and then finetuning over limited samples from the target domain. Yet, despite its popularity, this methodology has been shown to suffer in the presence of distributional shift -- specifically when the output spaces diverge. Previous work has focused on increasing model performance within this setting by identifying and classifying only the shared output classes between distributions. However, these methods are inherently limited as they ignore classes outside the shared class set, disregarding potential information relevant to the model transfer. This paper proposes a new process for few-shot transfer learning that selects and weighs classes from the source domain to optimize the transfer between domains. More concretely, we use Wasserstein distance to choose a set of source classes and their weights that minimize the distance between the source and target domain. To justify our proposed algorithm, we provide a generalization analysis of the performance of the learned classifier over the target domain and show that our method corresponds to a bound minimization algorithm. We empirically demonstrate the effectiveness of our approach (WaSS) by experimenting on several different datasets and presenting superior performance within various label shift settings, including the extreme case where the label spaces are disjoint.
MolCap-Arena: A Comprehensive Captioning Benchmark on Language-Enhanced Molecular Property Prediction
Nov 01, 2024



Bridging biomolecular modeling with natural language information, particularly through large language models (LLMs), has recently emerged as a promising interdisciplinary research area. LLMs, having been trained on large corpora of scientific documents, demonstrate significant potential in understanding and reasoning about biomolecules by providing enriched contextual and domain knowledge. However, the extent to which LLM-driven insights can improve performance on complex predictive tasks (e.g., toxicity) remains unclear. Further, the extent to which relevant knowledge can be extracted from LLMs also remains unknown. In this study, we present Molecule Caption Arena: the first comprehensive benchmark of LLM-augmented molecular property prediction. We evaluate over twenty LLMs, including both general-purpose and domain-specific molecule captioners, across diverse prediction tasks. To this goal, we introduce a novel, battle-based rating system. Our findings confirm the ability of LLM-extracted knowledge to enhance state-of-the-art molecular representations, with notable model-, prompt-, and dataset-specific variations. Code, resources, and data are available at github.com/Genentech/molcap-arena.
Geometry Informed Tokenization of Molecules for Language Model Generation
Aug 19, 2024We consider molecule generation in 3D space using language models (LMs), which requires discrete tokenization of 3D molecular geometries. Although tokenization of molecular graphs exists, that for 3D geometries is largely unexplored. Here, we attempt to bridge this gap by proposing the Geo2Seq, which converts molecular geometries into $SE(3)$-invariant 1D discrete sequences. Geo2Seq consists of canonical labeling and invariant spherical representation steps, which together maintain geometric and atomic fidelity in a format conducive to LMs. Our experiments show that, when coupled with Geo2Seq, various LMs excel in molecular geometry generation, especially in controlled generation tasks.
GLaD: Synergizing Molecular Graphs and Language Descriptors for Enhanced Power Conversion Efficiency Prediction in Organic Photovoltaic Devices
May 23, 2024This paper presents a novel approach for predicting Power Conversion Efficiency (PCE) of Organic Photovoltaic (OPV) devices, called GLaD: synergizing molecular Graphs and Language Descriptors for enhanced PCE prediction. Due to the lack of high-quality experimental data, we collect a dataset consisting of 500 pairs of OPV donor and acceptor molecules along with their corresponding PCE values, which we utilize as the training data for our predictive model. In this low-data regime, GLaD leverages properties learned from large language models (LLMs) pretrained on extensive scientific literature to enrich molecular structural representations, allowing for a multimodal representation of molecules. GLaD achieves precise predictions of PCE, thereby facilitating the synthesis of new OPV molecules with improved efficiency. Furthermore, GLaD showcases versatility, as it applies to a range of molecular property prediction tasks (BBBP, BACE, ClinTox, and SIDER), not limited to those concerning OPV materials. Especially, GLaD proves valuable for tasks in low-data regimes within the chemical space, as it enriches molecular representations by incorporating molecular property descriptions learned from large-scale pretraining. This capability is significant in real-world scientific endeavors like drug and material discovery, where access to comprehensive data is crucial for informed decision-making and efficient exploration of the chemical space.
$\textit{L+M-24}$: Building a Dataset for Language + Molecules @ ACL 2024
Feb 22, 2024Language-molecule models have emerged as an exciting direction for molecular discovery and understanding. However, training these models is challenging due to the scarcity of molecule-language pair datasets. At this point, datasets have been released which are 1) small and scraped from existing databases, 2) large but noisy and constructed by performing entity linking on the scientific literature, and 3) built by converting property prediction datasets to natural language using templates. In this document, we detail the $\textit{L+M-24}$ dataset, which has been created for the Language + Molecules Workshop shared task at ACL 2024. In particular, $\textit{L+M-24}$ is designed to focus on three key benefits of natural language in molecule design: compositionality, functionality, and abstraction.
ChemReasoner: Heuristic Search over a Large Language Model's Knowledge Space using Quantum-Chemical Feedback
Feb 21, 2024



The discovery of new catalysts is essential for the design of new and more efficient chemical processes in order to transition to a sustainable future. We introduce an AI-guided computational screening framework unifying linguistic reasoning with quantum-chemistry based feedback from 3D atomistic representations. Our approach formulates catalyst discovery as an uncertain environment where an agent actively searches for highly effective catalysts via the iterative combination of large language model (LLM)-derived hypotheses and atomistic graph neural network (GNN)-derived feedback. Identified catalysts in intermediate search steps undergo structural evaluation based on spatial orientation, reaction pathways, and stability. Scoring functions based on adsorption energies and barriers steer the exploration in the LLM's knowledge space toward energetically favorable, high-efficiency catalysts. We introduce planning methods that automatically guide the exploration without human input, providing competitive performance against expert-enumerated chemical descriptor-based implementations. By integrating language-guided reasoning with computational chemistry feedback, our work pioneers AI-accelerated, trustworthy catalyst discovery.
Defining a New NLP Playground
Oct 31, 2023The recent explosion of performance of large language models (LLMs) has changed the field of Natural Language Processing (NLP) more abruptly and seismically than any other shift in the field's 80-year history. This has resulted in concerns that the field will become homogenized and resource-intensive. The new status quo has put many academic researchers, especially PhD students, at a disadvantage. This paper aims to define a new NLP playground by proposing 20+ PhD-dissertation-worthy research directions, covering theoretical analysis, new and challenging problems, learning paradigms, and interdisciplinary applications.