 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePRISM: A Unified Framework for Post-Training LLMs Without Verifiable Rewards
Jan 08, 2026Current techniques for post-training Large Language Models (LLMs) rely either on costly human supervision or on external verifiers to boost performance on tasks such as mathematical reasoning and code generation. However, as LLMs improve their problem-solving, any further improvement will potentially require high-quality solutions to difficult problems that are not available to humans. As a result, learning from unlabeled data is becoming increasingly attractive in the research community. Existing methods extract learning signal from a model's consistency, either by majority voting or by converting the model's internal confidence into reward. Although internal consistency metric such as entropy or self-certainty require no human intervention, as we show in this work, these are unreliable signals for large-scale and long-term training. To address the unreliability, we propose PRISM, a unified training framework that uses a Process Reward Model (PRM) to guide learning alongside model's internal confidence in the absence of ground-truth labels. We show that effectively combining PRM with self-certainty can lead to both stable training and better test-time performance, and also keep the model's internal confidence in check.
Learning Disentangled Equivariant Representation for Explicitly Controllable 3D Molecule Generation
Dec 19, 2024We consider the conditional generation of 3D drug-like molecules with \textit{explicit control} over molecular properties such as drug-like properties (e.g., Quantitative Estimate of Druglikeness or Synthetic Accessibility score) and effectively binding to specific protein sites. To tackle this problem, we propose an E(3)-equivariant Wasserstein autoencoder and factorize the latent space of our generative model into two disentangled aspects: molecular properties and the remaining structural context of 3D molecules. Our model ensures explicit control over these molecular attributes while maintaining equivariance of coordinate representation and invariance of data likelihood. Furthermore, we introduce a novel alignment-based coordinate loss to adapt equivariant networks for auto-regressive de-novo 3D molecule generation from scratch. Extensive experiments validate our model's effectiveness on property-guided and context-guided molecule generation, both for de-novo 3D molecule design and structure-based drug discovery against protein targets.
Geometry Informed Tokenization of Molecules for Language Model Generation
Aug 19, 2024We consider molecule generation in 3D space using language models (LMs), which requires discrete tokenization of 3D molecular geometries. Although tokenization of molecular graphs exists, that for 3D geometries is largely unexplored. Here, we attempt to bridge this gap by proposing the Geo2Seq, which converts molecular geometries into $SE(3)$-invariant 1D discrete sequences. Geo2Seq consists of canonical labeling and invariant spherical representation steps, which together maintain geometric and atomic fidelity in a format conducive to LMs. Our experiments show that, when coupled with Geo2Seq, various LMs excel in molecular geometry generation, especially in controlled generation tasks.
Artificial Intelligence for Science in Quantum, Atomistic, and Continuum Systems
Jul 17, 2023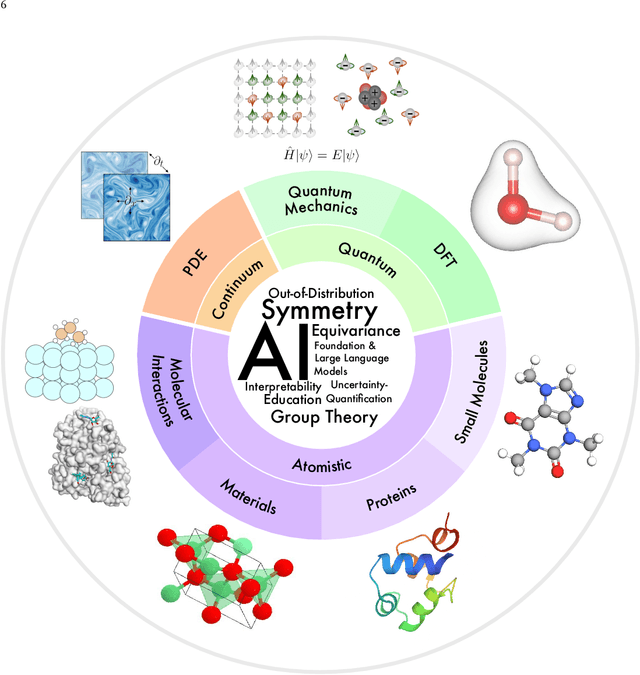
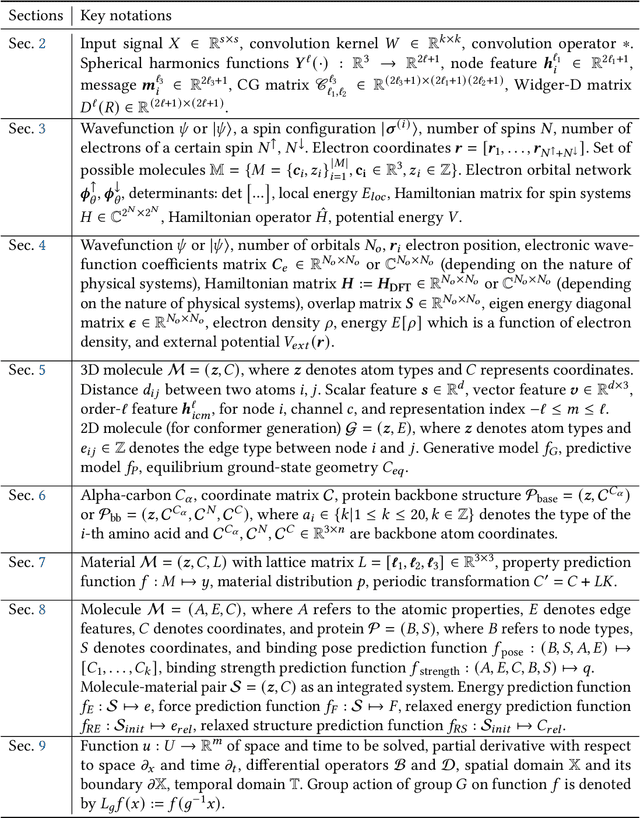
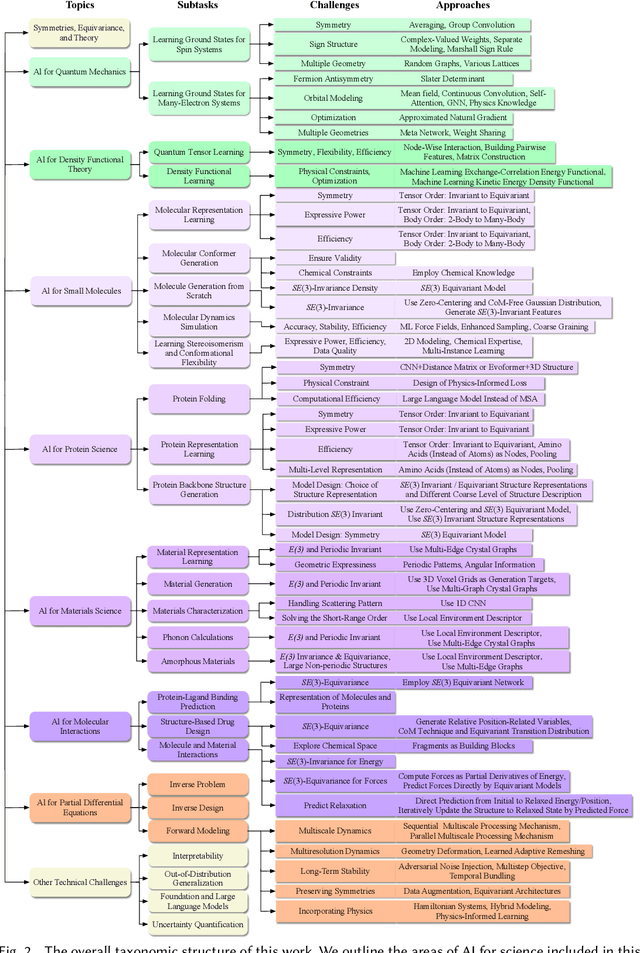

Advances in artificial intelligence (AI) are fueling a new paradigm of discoveries in natural sciences. Today, AI has started to advance natural sciences by improving, accelerating, and enabling our understanding of natural phenomena at a wide range of spatial and temporal scales, giving rise to a new area of research known as AI for science (AI4Science). Being an emerging research paradigm, AI4Science is unique in that it is an enormous and highly interdisciplinary area. Thus, a unified and technical treatment of this field is needed yet challenging. This paper aims to provide a technically thorough account of a subarea of AI4Science; namely, AI for quantum, atomistic, and continuum systems. These areas aim at understanding the physical world from the subatomic (wavefunctions and electron density), atomic (molecules, proteins, materials, and interactions), to macro (fluids, climate, and subsurface) scales and form an important subarea of AI4Science. A unique advantage of focusing on these areas is that they largely share a common set of challenges, thereby allowing a unified and foundational treatment. A key common challenge is how to capture physics first principles, especially symmetries, in natural systems by deep learning methods. We provide an in-depth yet intuitive account of techniques to achieve equivariance to symmetry transformations. We also discuss other common technical challenges, including explainability, out-of-distribution generalization, knowledge transfer with foundation and large language models, and uncertainty quantification. To facilitate learning and education, we provide categorized lists of resources that we found to be useful. We strive to be thorough and unified and hope this initial effort may trigger more community interests and efforts to further advance AI4Science.
Towards Symmetry-Aware Generation of Periodic Materials
Jul 06, 2023



We consider the problem of generating periodic materials with deep models. While symmetry-aware molecule generation has been studied extensively, periodic materials possess different symmetries, which have not been completely captured by existing methods. In this work, we propose SyMat, a novel material generation approach that can capture physical symmetries of periodic material structures. SyMat generates atom types and lattices of materials through generating atom type sets, lattice lengths and lattice angles with a variational auto-encoder model. In addition, SyMat employs a score-based diffusion model to generate atom coordinates of materials, in which a novel symmetry-aware probabilistic model is used in the coordinate diffusion process. We show that SyMat is theoretically invariant to all symmetry transformations on materials and demonstrate that SyMat achieves promising performance on random generation and property optimization tasks.
Efficient Approximations of Complete Interatomic Potentials for Crystal Property Prediction
Jun 26, 2023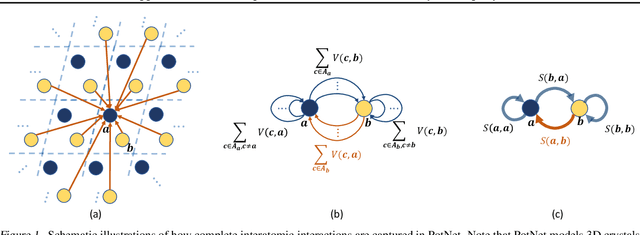
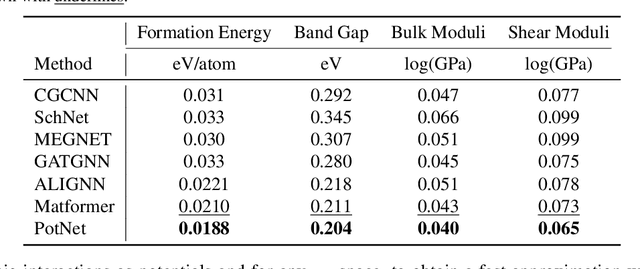
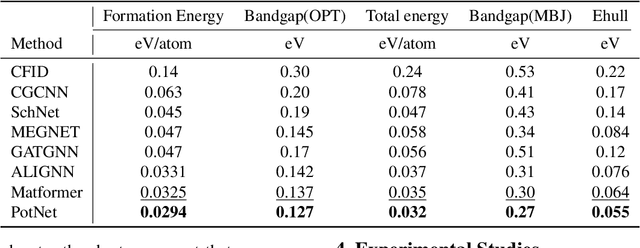
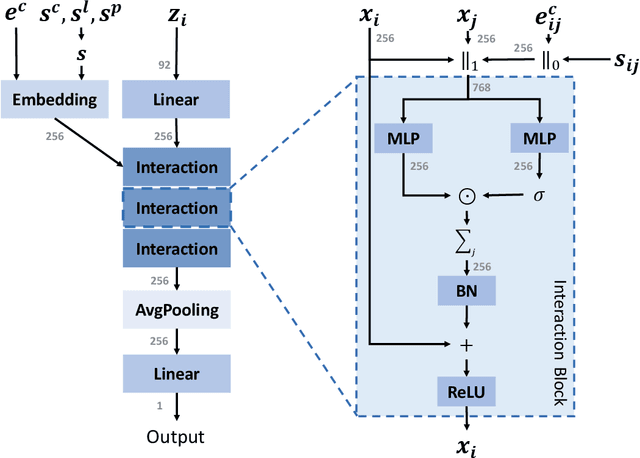
We study property prediction for crystal materials. A crystal structure consists of a minimal unit cell that is repeated infinitely in 3D space. How to accurately represent such repetitive structures in machine learning models remains unresolved. Current methods construct graphs by establishing edges only between nearby nodes, thereby failing to faithfully capture infinite repeating patterns and distant interatomic interactions. In this work, we propose several innovations to overcome these limitations. First, we propose to model physics-principled interatomic potentials directly instead of only using distances as in many existing methods. These potentials include the Coulomb potential, London dispersion potential, and Pauli repulsion potential. Second, we model the complete set of potentials among all atoms, instead of only between nearby atoms as in existing methods. This is enabled by our approximations of infinite potential summations with provable error bounds. We further develop efficient algorithms to compute the approximations. Finally, we propose to incorporate our computations of complete interatomic potentials into message passing neural networks for representation learning. We perform experiments on the JARVIS and Materials Project benchmarks for evaluation. Results show that the use of interatomic potentials and complete interatomic potentials leads to consistent performance improvements with reasonable computational costs. Our code is publicly available as part of the AIRS library (https://github.com/divelab/AIRS/tree/main/OpenMat/PotNet).
QH9: A Quantum Hamiltonian Prediction Benchmark for QM9 Molecules
Jun 15, 2023



Supervised machine learning approaches have been increasingly used in accelerating electronic structure prediction as surrogates of first-principle computational methods, such as density functional theory (DFT). While numerous quantum chemistry datasets focus on chemical properties and atomic forces, the ability to achieve accurate and efficient prediction of the Hamiltonian matrix is highly desired, as it is the most important and fundamental physical quantity that determines the quantum states of physical systems and chemical properties. In this work, we generate a new Quantum Hamiltonian dataset, named as QH9, to provide precise Hamiltonian matrices for 2,399 molecular dynamics trajectories and 130,831 stable molecular geometries, based on the QM9 dataset. By designing benchmark tasks with various molecules, we show that current machine learning models have the capacity to predict Hamiltonian matrices for arbitrary molecules. Both the QH9 dataset and the baseline models are provided to the community through an open-source benchmark, which can be highly valuable for developing machine learning methods and accelerating molecular and materials design for scientific and technological applications. Our benchmark is publicly available at https://github.com/divelab/AIRS/tree/main/OpenDFT/QHBench.
Graph Structure and Feature Extrapolation for Out-of-Distribution Generalization
Jun 13, 2023



Out-of-distribution (OOD) generalization deals with the prevalent learning scenario where test distribution shifts from training distribution. With rising application demands and inherent complexity, graph OOD problems call for specialized solutions. While data-centric methods exhibit performance enhancements on many generic machine learning tasks, there is a notable absence of data augmentation methods tailored for graph OOD generalization. In this work, we propose to achieve graph OOD generalization with the novel design of non-Euclidean-space linear extrapolation. The proposed augmentation strategy extrapolates both structure and feature spaces to generate OOD graph data. Our design tailors OOD samples for specific shifts without corrupting underlying causal mechanisms. Theoretical analysis and empirical results evidence the effectiveness of our method in solving target shifts, showing substantial and constant improvements across various graph OOD tasks.
Joint Learning of Label and Environment Causal Independence for Graph Out-of-Distribution Generalization
Jun 08, 2023
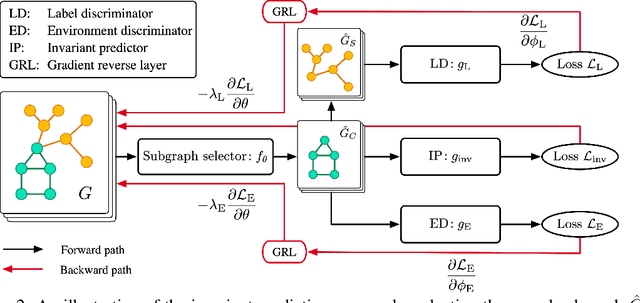
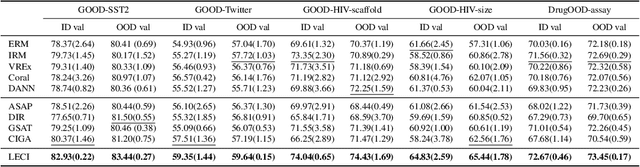

We tackle the problem of graph out-of-distribution (OOD) generalization. Existing graph OOD algorithms either rely on restricted assumptions or fail to exploit environment information in training data. In this work, we propose to simultaneously incorporate label and environment causal independence (LECI) to fully make use of label and environment information, thereby addressing the challenges faced by prior methods on identifying causal and invariant subgraphs. We further develop an adversarial training strategy to jointly optimize these two properties for causal subgraph discovery with theoretical guarantees. Extensive experiments and analysis show that LECI significantly outperforms prior methods on both synthetic and real-world datasets, establishing LECI as a practical and effective solution for graph OOD generalization.
Generating 3D Molecules for Target Protein Binding
Apr 19, 2022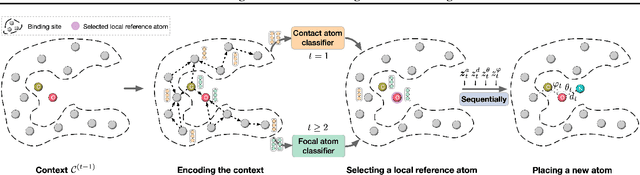
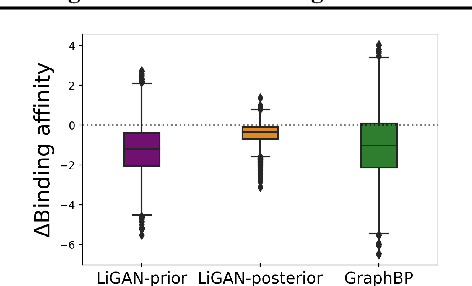
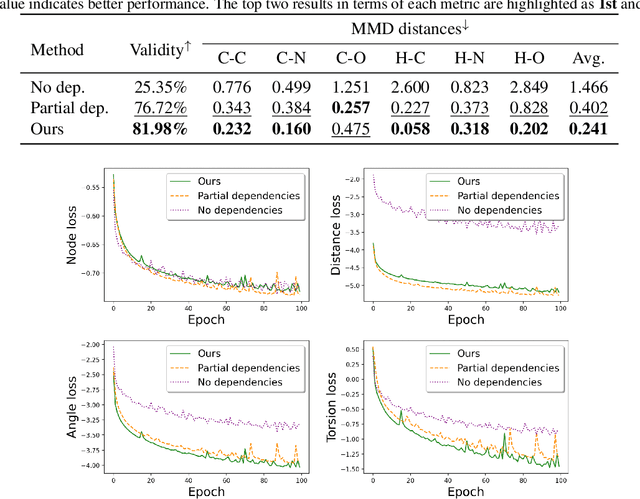
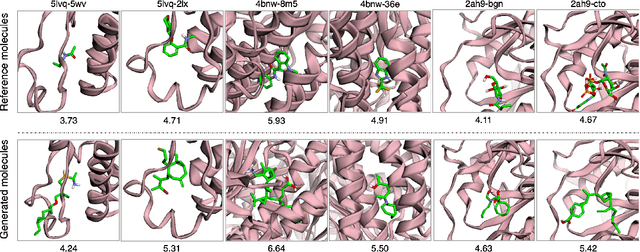
A fundamental problem in drug discovery is to design molecules that bind to specific proteins. To tackle this problem using machine learning methods, here we propose a novel and effective framework, known as GraphBP, to generate 3D molecules that bind to given proteins by placing atoms of specific types and locations to the given binding site one by one. In particular, at each step, we first employ a 3D graph neural network to obtain geometry-aware and chemically informative representations from the intermediate contextual information. Such context includes the given binding site and atoms placed in the previous steps. Second, to preserve the desirable equivariance property, we select a local reference atom according to the designed auxiliary classifiers and then construct a local spherical coordinate system. Finally, to place a new atom, we generate its atom type and relative location w.r.t. the constructed local coordinate system via a flow model. We also consider generating the variables of interest sequentially to capture the underlying dependencies among them. Experiments demonstrate that our GraphBP is effective to generate 3D molecules with binding ability to target protein binding sites. Our implementation is available at https://github.com/divelab/GraphBP.