 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeArtificial Intelligence for Science in Quantum, Atomistic, and Continuum Systems
Jul 17, 2023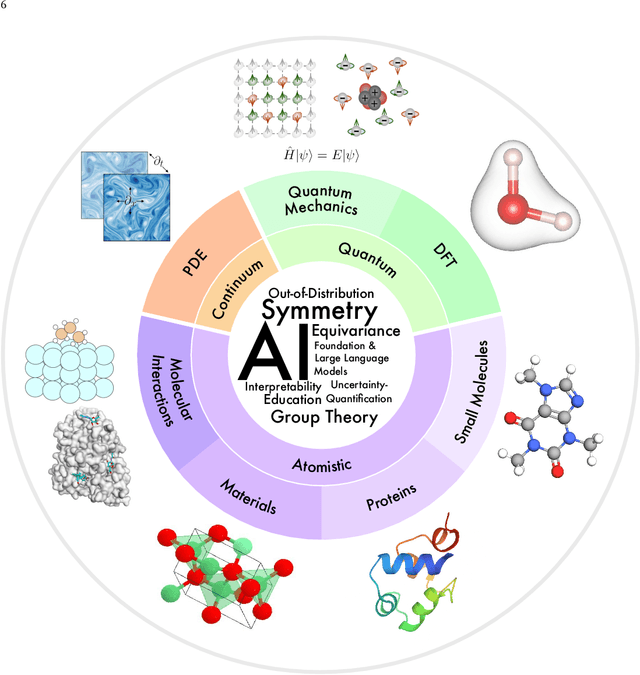
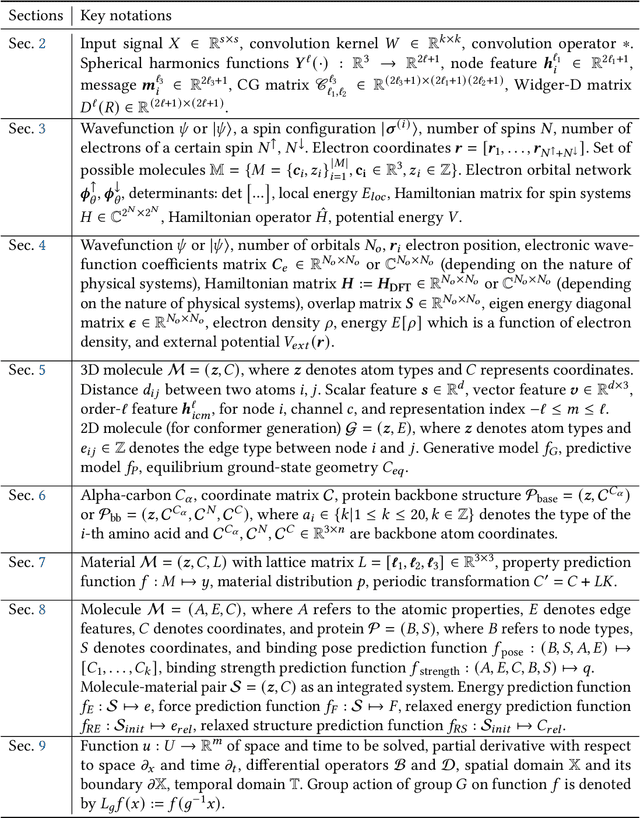
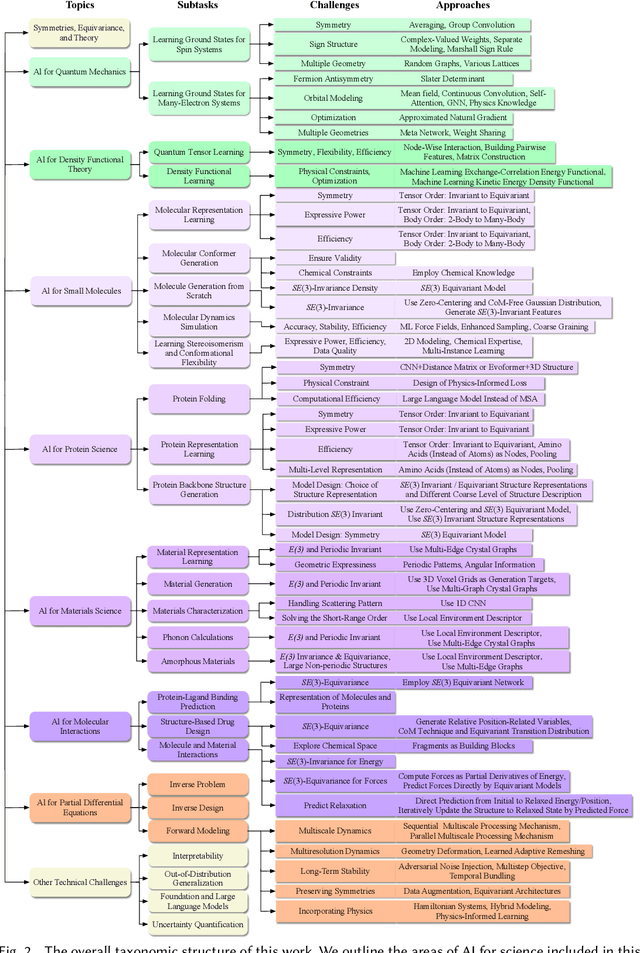

Advances in artificial intelligence (AI) are fueling a new paradigm of discoveries in natural sciences. Today, AI has started to advance natural sciences by improving, accelerating, and enabling our understanding of natural phenomena at a wide range of spatial and temporal scales, giving rise to a new area of research known as AI for science (AI4Science). Being an emerging research paradigm, AI4Science is unique in that it is an enormous and highly interdisciplinary area. Thus, a unified and technical treatment of this field is needed yet challenging. This paper aims to provide a technically thorough account of a subarea of AI4Science; namely, AI for quantum, atomistic, and continuum systems. These areas aim at understanding the physical world from the subatomic (wavefunctions and electron density), atomic (molecules, proteins, materials, and interactions), to macro (fluids, climate, and subsurface) scales and form an important subarea of AI4Science. A unique advantage of focusing on these areas is that they largely share a common set of challenges, thereby allowing a unified and foundational treatment. A key common challenge is how to capture physics first principles, especially symmetries, in natural systems by deep learning methods. We provide an in-depth yet intuitive account of techniques to achieve equivariance to symmetry transformations. We also discuss other common technical challenges, including explainability, out-of-distribution generalization, knowledge transfer with foundation and large language models, and uncertainty quantification. To facilitate learning and education, we provide categorized lists of resources that we found to be useful. We strive to be thorough and unified and hope this initial effort may trigger more community interests and efforts to further advance AI4Science.
QH9: A Quantum Hamiltonian Prediction Benchmark for QM9 Molecules
Jun 15, 2023



Supervised machine learning approaches have been increasingly used in accelerating electronic structure prediction as surrogates of first-principle computational methods, such as density functional theory (DFT). While numerous quantum chemistry datasets focus on chemical properties and atomic forces, the ability to achieve accurate and efficient prediction of the Hamiltonian matrix is highly desired, as it is the most important and fundamental physical quantity that determines the quantum states of physical systems and chemical properties. In this work, we generate a new Quantum Hamiltonian dataset, named as QH9, to provide precise Hamiltonian matrices for 2,399 molecular dynamics trajectories and 130,831 stable molecular geometries, based on the QM9 dataset. By designing benchmark tasks with various molecules, we show that current machine learning models have the capacity to predict Hamiltonian matrices for arbitrary molecules. Both the QH9 dataset and the baseline models are provided to the community through an open-source benchmark, which can be highly valuable for developing machine learning methods and accelerating molecular and materials design for scientific and technological applications. Our benchmark is publicly available at https://github.com/divelab/AIRS/tree/main/OpenDFT/QHBench.