 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAgentA/B: Automated and Scalable Web A/BTesting with Interactive LLM Agents
Apr 13, 2025



A/B testing experiment is a widely adopted method for evaluating UI/UX design decisions in modern web applications. Yet, traditional A/B testing remains constrained by its dependence on the large-scale and live traffic of human participants, and the long time of waiting for the testing result. Through formative interviews with six experienced industry practitioners, we identified critical bottlenecks in current A/B testing workflows. In response, we present AgentA/B, a novel system that leverages Large Language Model-based autonomous agents (LLM Agents) to automatically simulate user interaction behaviors with real webpages. AgentA/B enables scalable deployment of LLM agents with diverse personas, each capable of navigating the dynamic webpage and interactively executing multi-step interactions like search, clicking, filtering, and purchasing. In a demonstrative controlled experiment, we employ AgentA/B to simulate a between-subject A/B testing with 1,000 LLM agents Amazon.com, and compare agent behaviors with real human shopping behaviors at a scale. Our findings suggest AgentA/B can emulate human-like behavior patterns.
Beyond Believability: Accurate Human Behavior Simulation with Fine-Tuned LLMs
Mar 27, 2025Recent research shows that LLMs can simulate ``believable'' human behaviors to power LLM agents via prompt-only methods. In this work, we focus on evaluating and improving LLM's objective ``accuracy'' rather than the subjective ``believability'' in the web action generation task, leveraging a large-scale, real-world dataset collected from online shopping human actions. We present the first comprehensive quantitative evaluation of state-of-the-art LLMs (e.g., DeepSeek-R1, Llama, and Claude) on the task of web action generation. Our results show that fine-tuning LLMs on real-world behavioral data substantially improves their ability to generate actions compared to prompt-only methods. Furthermore, incorporating synthesized reasoning traces into model training leads to additional performance gains, demonstrating the value of explicit rationale in behavior modeling. This work establishes a new benchmark for evaluating LLMs in behavior simulation and offers actionable insights into how real-world action data and reasoning augmentation can enhance the fidelity of LLM agents.
Reasoning with Graphs: Structuring Implicit Knowledge to Enhance LLMs Reasoning
Jan 14, 2025Large language models (LLMs) have demonstrated remarkable success across a wide range of tasks; however, they still encounter challenges in reasoning tasks that require understanding and inferring relationships between distinct pieces of information within text sequences. This challenge is particularly pronounced in tasks involving multi-step processes, such as logical reasoning and multi-hop question answering, where understanding implicit relationships between entities and leveraging multi-hop connections in the given context are crucial. Graphs, as fundamental data structures, explicitly represent pairwise relationships between entities, thereby offering the potential to enhance LLMs' reasoning capabilities. External graphs have proven effective in supporting LLMs across multiple tasks. However, in many reasoning tasks, no pre-existing graph structure is provided. Can we structure implicit knowledge derived from context into graphs to assist LLMs in reasoning? In this paper, we propose Reasoning with Graphs (RwG) by first constructing explicit graphs from the context and then leveraging these graphs to enhance LLM reasoning performance on reasoning tasks. Extensive experiments demonstrate the effectiveness of the proposed method in improving both logical reasoning and multi-hop question answering tasks.
SimRAG: Self-Improving Retrieval-Augmented Generation for Adapting Large Language Models to Specialized Domains
Oct 23, 2024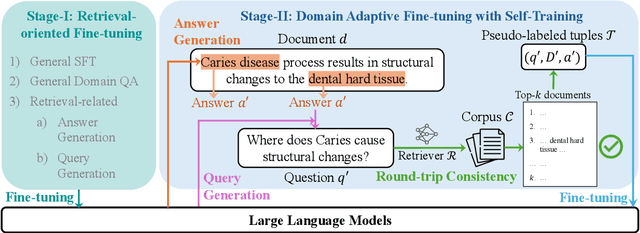
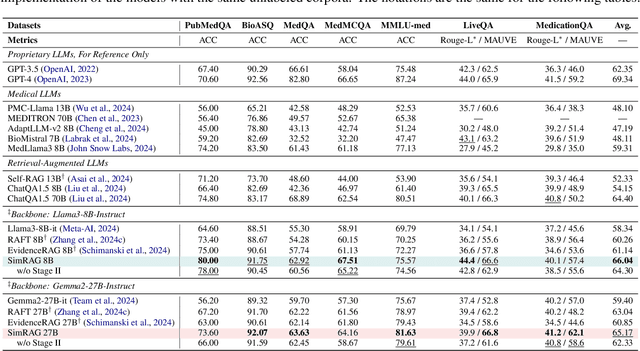
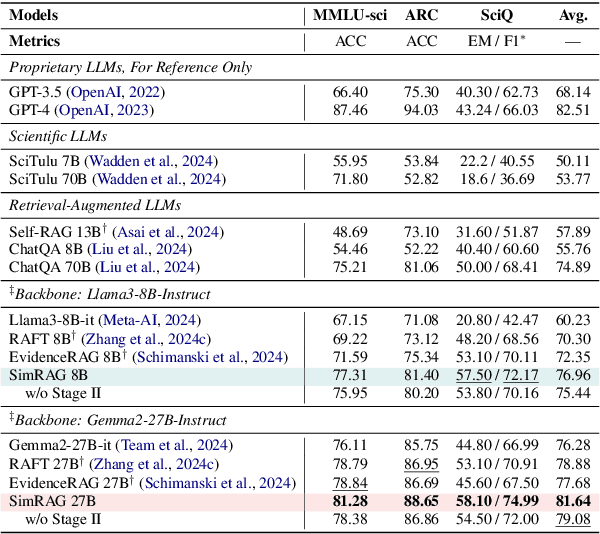
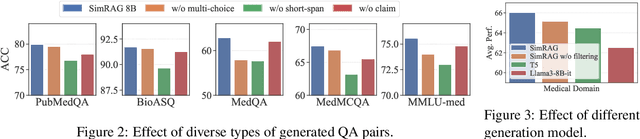
Retrieval-augmented generation (RAG) enhances the question-answering (QA) abilities of large language models (LLMs) by integrating external knowledge. However, adapting general-purpose RAG systems to specialized fields such as science and medicine poses unique challenges due to distribution shifts and limited access to domain-specific data. To tackle this, we propose SimRAG, a self-training approach that equips the LLM with joint capabilities of question answering and question generation for domain adaptation. Our method first fine-tunes the LLM on instruction-following, question-answering, and search-related data. Then, it prompts the same LLM to generate diverse domain-relevant questions from unlabeled corpora, with an additional filtering strategy to retain high-quality synthetic examples. By leveraging these synthetic examples, the LLM can improve their performance on domain-specific RAG tasks. Experiments on 11 datasets, spanning two backbone sizes and three domains, demonstrate that SimRAG outperforms baselines by 1.2\%--8.6\%.
Exploring Query Understanding for Amazon Product Search
Aug 05, 2024



Online shopping platforms, such as Amazon, offer services to billions of people worldwide. Unlike web search or other search engines, product search engines have their unique characteristics, primarily featuring short queries which are mostly a combination of product attributes and structured product search space. The uniqueness of product search underscores the crucial importance of the query understanding component. However, there are limited studies focusing on exploring this impact within real-world product search engines. In this work, we aim to bridge this gap by conducting a comprehensive study and sharing our year-long journey investigating how the query understanding service impacts Amazon Product Search. Firstly, we explore how query understanding-based ranking features influence the ranking process. Next, we delve into how the query understanding system contributes to understanding the performance of a ranking model. Building on the insights gained from our study on the evaluation of the query understanding-based ranking model, we propose a query understanding-based multi-task learning framework for ranking. We present our studies and investigations using the real-world system on Amazon Search.
SineNet: Learning Temporal Dynamics in Time-Dependent Partial Differential Equations
Mar 28, 2024



We consider using deep neural networks to solve time-dependent partial differential equations (PDEs), where multi-scale processing is crucial for modeling complex, time-evolving dynamics. While the U-Net architecture with skip connections is commonly used by prior studies to enable multi-scale processing, our analysis shows that the need for features to evolve across layers results in temporally misaligned features in skip connections, which limits the model's performance. To address this limitation, we propose SineNet, consisting of multiple sequentially connected U-shaped network blocks, referred to as waves. In SineNet, high-resolution features are evolved progressively through multiple stages, thereby reducing the amount of misalignment within each stage. We furthermore analyze the role of skip connections in enabling both parallel and sequential processing of multi-scale information. Our method is rigorously tested on multiple PDE datasets, including the Navier-Stokes equations and shallow water equations, showcasing the advantages of our proposed approach over conventional U-Nets with a comparable parameter budget. We further demonstrate that increasing the number of waves in SineNet while maintaining the same number of parameters leads to a monotonically improved performance. The results highlight the effectiveness of SineNet and the potential of our approach in advancing the state-of-the-art in neural PDE solver design. Our code is available as part of AIRS (https://github.com/divelab/AIRS).
Genetic InfoMax: Exploring Mutual Information Maximization in High-Dimensional Imaging Genetics Studies
Sep 26, 2023Genome-wide association studies (GWAS) are used to identify relationships between genetic variations and specific traits. When applied to high-dimensional medical imaging data, a key step is to extract lower-dimensional, yet informative representations of the data as traits. Representation learning for imaging genetics is largely under-explored due to the unique challenges posed by GWAS in comparison to typical visual representation learning. In this study, we tackle this problem from the mutual information (MI) perspective by identifying key limitations of existing methods. We introduce a trans-modal learning framework Genetic InfoMax (GIM), including a regularized MI estimator and a novel genetics-informed transformer to address the specific challenges of GWAS. We evaluate GIM on human brain 3D MRI data and establish standardized evaluation protocols to compare it to existing approaches. Our results demonstrate the effectiveness of GIM and a significantly improved performance on GWAS.
Artificial Intelligence for Science in Quantum, Atomistic, and Continuum Systems
Jul 17, 2023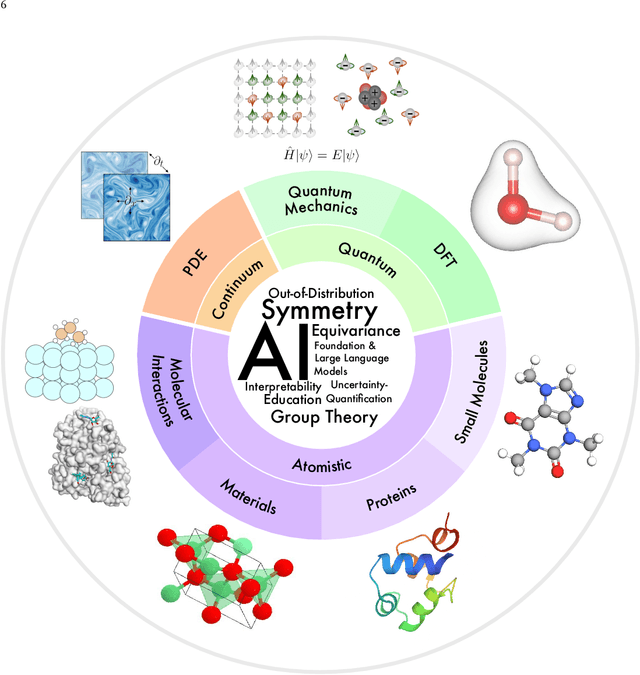
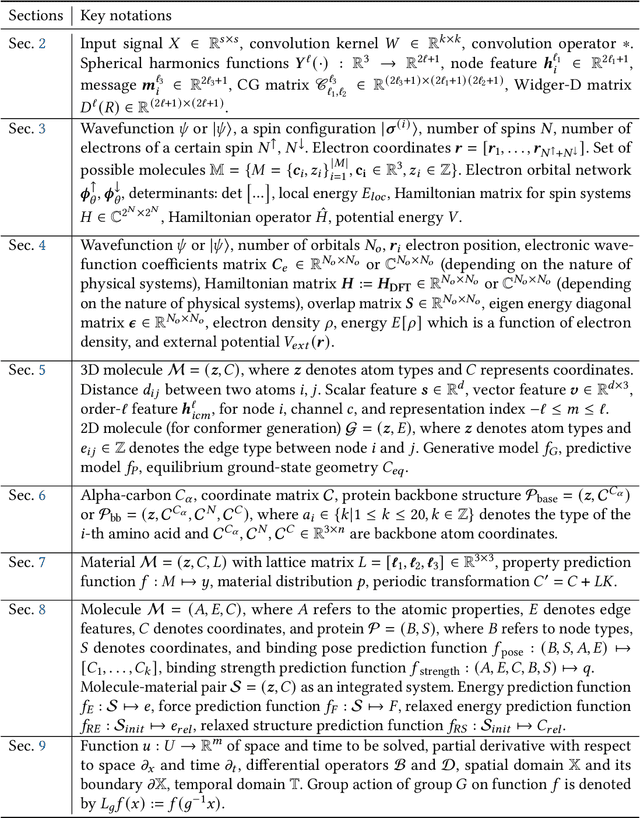
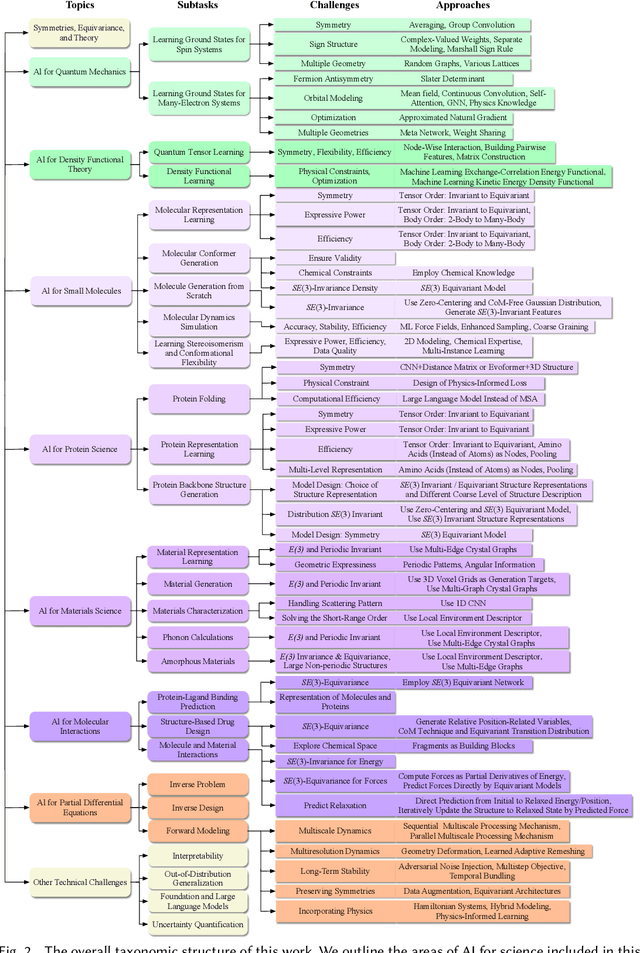

Advances in artificial intelligence (AI) are fueling a new paradigm of discoveries in natural sciences. Today, AI has started to advance natural sciences by improving, accelerating, and enabling our understanding of natural phenomena at a wide range of spatial and temporal scales, giving rise to a new area of research known as AI for science (AI4Science). Being an emerging research paradigm, AI4Science is unique in that it is an enormous and highly interdisciplinary area. Thus, a unified and technical treatment of this field is needed yet challenging. This paper aims to provide a technically thorough account of a subarea of AI4Science; namely, AI for quantum, atomistic, and continuum systems. These areas aim at understanding the physical world from the subatomic (wavefunctions and electron density), atomic (molecules, proteins, materials, and interactions), to macro (fluids, climate, and subsurface) scales and form an important subarea of AI4Science. A unique advantage of focusing on these areas is that they largely share a common set of challenges, thereby allowing a unified and foundational treatment. A key common challenge is how to capture physics first principles, especially symmetries, in natural systems by deep learning methods. We provide an in-depth yet intuitive account of techniques to achieve equivariance to symmetry transformations. We also discuss other common technical challenges, including explainability, out-of-distribution generalization, knowledge transfer with foundation and large language models, and uncertainty quantification. To facilitate learning and education, we provide categorized lists of resources that we found to be useful. We strive to be thorough and unified and hope this initial effort may trigger more community interests and efforts to further advance AI4Science.
Task-Agnostic Graph Explanations
Feb 16, 2022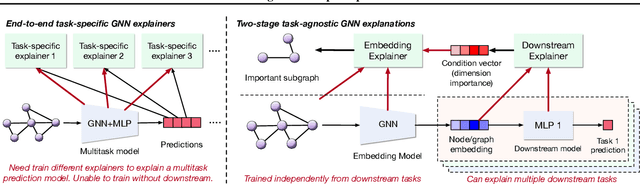
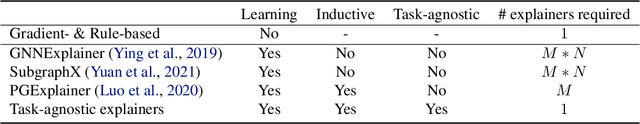
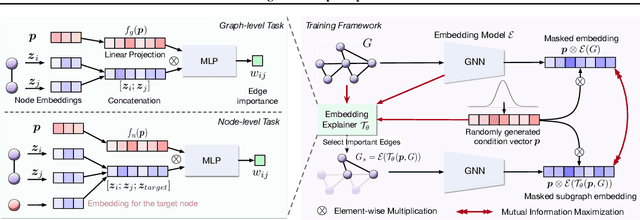
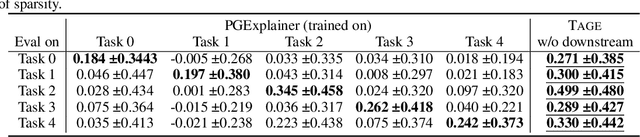
Graph Neural Networks (GNNs) have emerged as powerful tools to encode graph structured data. Due to their broad applications, there is an increasing need to develop tools to explain how GNNs make decisions given graph structured data. Existing learning-based GNN explanation approaches are task-specific in training and hence suffer from crucial drawbacks. Specifically, they are incapable of producing explanations for a multitask prediction model with a single explainer. They are also unable to provide explanations in cases where the GNN is trained in a self-supervised manner, and the resulting representations are used in future downstream tasks. To address these limitations, we propose a Task-Agnostic GNN Explainer (TAGE) trained under self-supervision with no knowledge of downstream tasks. TAGE enables the explanation of GNN embedding models without downstream tasks and allows efficient explanation of multitask models. Our extensive experiments show that TAGE can significantly speed up the explanation efficiency by using the same model to explain predictions for multiple downstream tasks while achieving explanation quality as good as or even better than current state-of-the-art GNN explanation approaches.
Self-Supervised Representation Learning via Latent Graph Prediction
Feb 16, 2022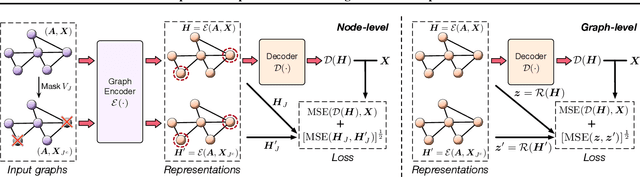
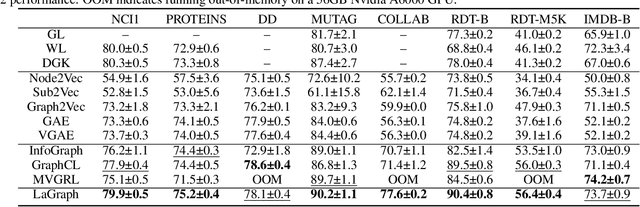
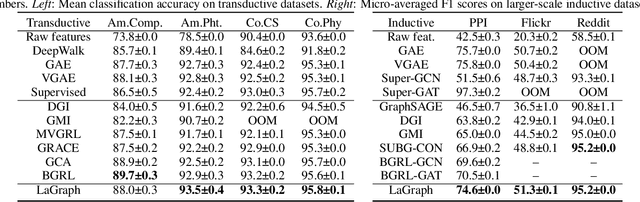
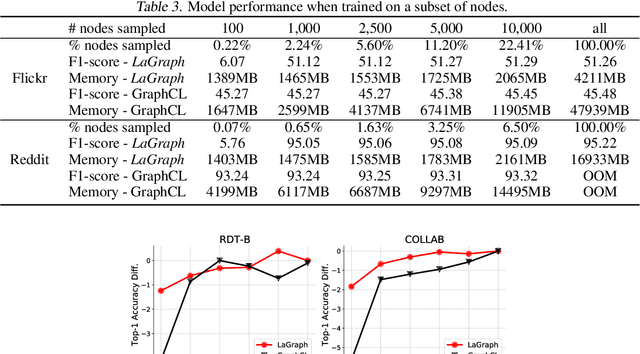
Self-supervised learning (SSL) of graph neural networks is emerging as a promising way of leveraging unlabeled data. Currently, most methods are based on contrastive learning adapted from the image domain, which requires view generation and a sufficient number of negative samples. In contrast, existing predictive models do not require negative sampling, but lack theoretical guidance on the design of pretext training tasks. In this work, we propose the LaGraph, a theoretically grounded predictive SSL framework based on latent graph prediction. Learning objectives of LaGraph are derived as self-supervised upper bounds to objectives for predicting unobserved latent graphs. In addition to its improved performance, LaGraph provides explanations for recent successes of predictive models that include invariance-based objectives. We provide theoretical analysis comparing LaGraph to related methods in different domains. Our experimental results demonstrate the superiority of LaGraph in performance and the robustness to decreasing of training sample size on both graph-level and node-level tasks.