 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGenetic InfoMax: Exploring Mutual Information Maximization in High-Dimensional Imaging Genetics Studies
Sep 26, 2023Genome-wide association studies (GWAS) are used to identify relationships between genetic variations and specific traits. When applied to high-dimensional medical imaging data, a key step is to extract lower-dimensional, yet informative representations of the data as traits. Representation learning for imaging genetics is largely under-explored due to the unique challenges posed by GWAS in comparison to typical visual representation learning. In this study, we tackle this problem from the mutual information (MI) perspective by identifying key limitations of existing methods. We introduce a trans-modal learning framework Genetic InfoMax (GIM), including a regularized MI estimator and a novel genetics-informed transformer to address the specific challenges of GWAS. We evaluate GIM on human brain 3D MRI data and establish standardized evaluation protocols to compare it to existing approaches. Our results demonstrate the effectiveness of GIM and a significantly improved performance on GWAS.
Deep Learning based Early Detection and Grading of Diabetic Retinopathy Using Retinal Fundus Images
Dec 27, 2018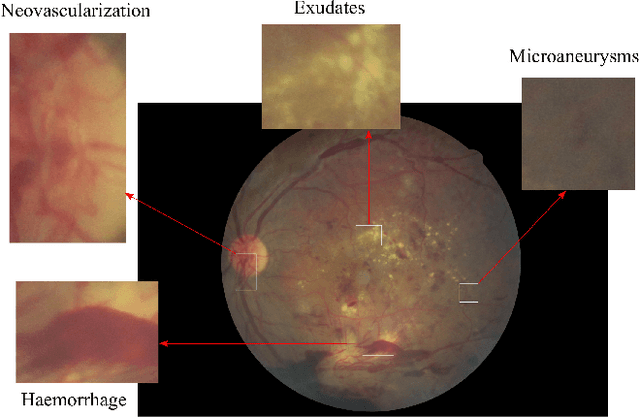
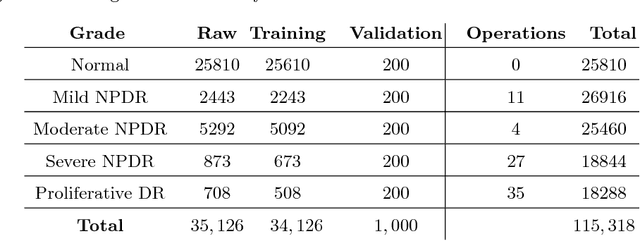
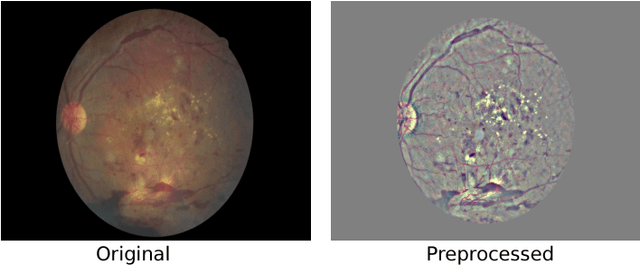
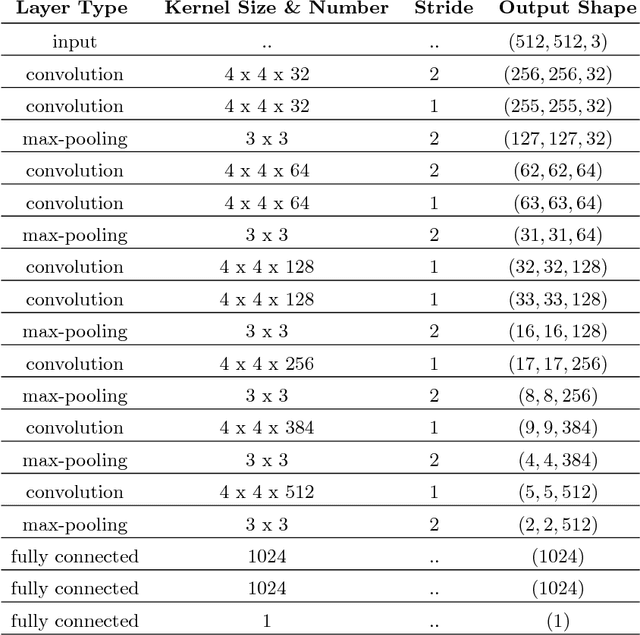
Diabetic Retinopathy (DR) is a constantly deteriorating disease, being one of the leading causes of vision impairment and blindness. Subtle distinction among different grades and existence of many significant small features make the task of recognition very challenging. In addition, the present approach of retinopathy detection is a very laborious and time-intensive task, which heavily relies on the skill of a physician. Automated detection of diabetic retinopathy is essential to tackle these problems. Early-stage detection of diabetic retinopathy is also very important for diagnosis, which can prevent blindness with proper treatment. In this paper, we developed a novel deep convolutional neural network, which performs the early-stage detection by identifying all microaneurysms (MAs), the first signs of DR, along with correctly assigning labels to retinal fundus images which are graded into five categories. We have tested our network on the largest publicly available Kaggle diabetic retinopathy dataset, and achieved 0.851 quadratic weighted kappa score and 0.844 AUC score, which achieves the state-of-the-art performance on severity grading. In the early-stage detection, we have achieved a sensitivity of 98% and specificity of above 94%, which demonstrates the effectiveness of our proposed method. Our proposed architecture is at the same time very simple and efficient with respect to computational time and space are concerned.
DEEPGONET: Multi-label Prediction of GO Annotation for Protein from Sequence Using Cascaded Convolutional and Recurrent Network
Oct 31, 2018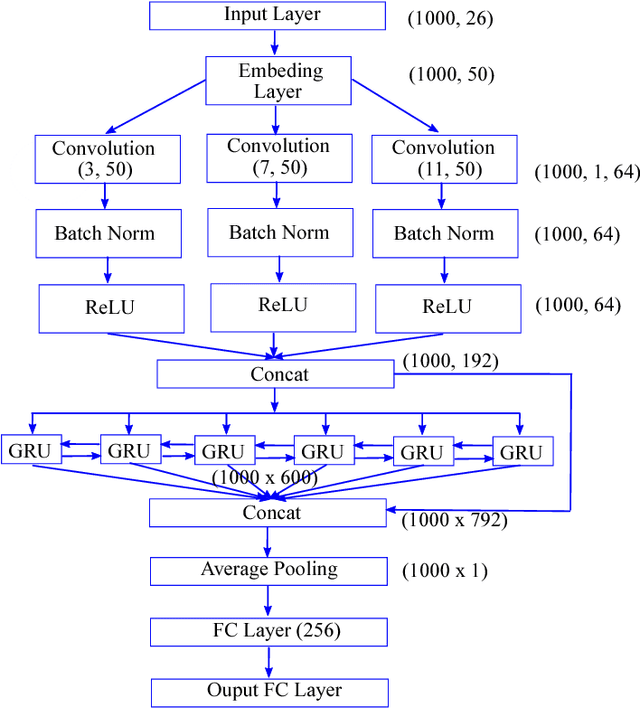
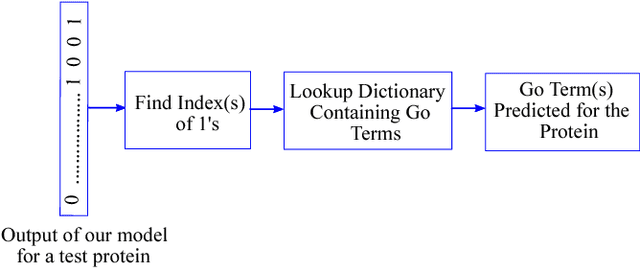
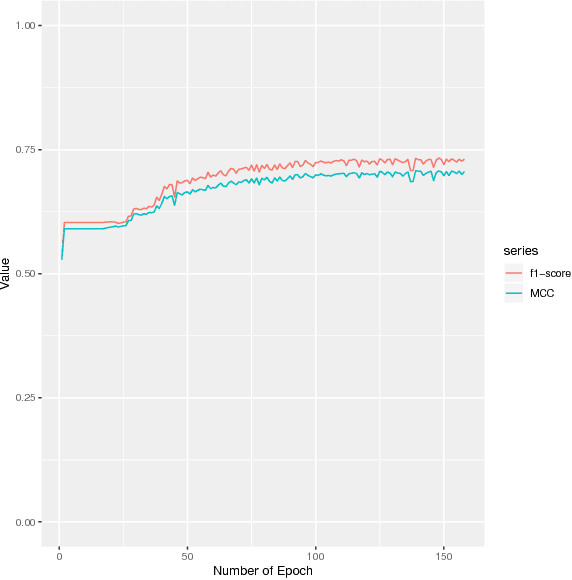
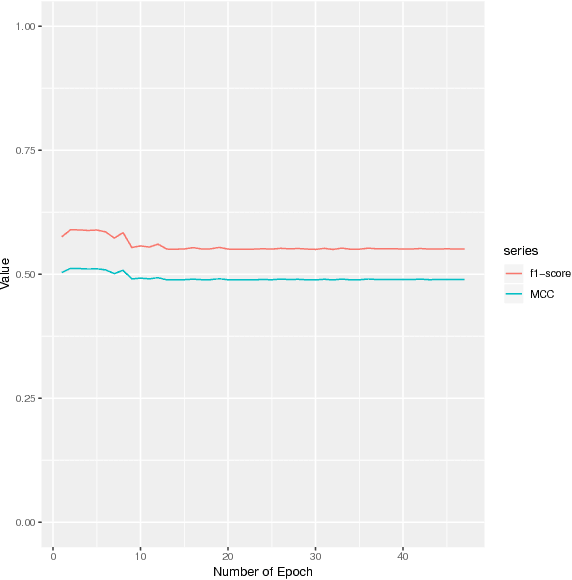
The present gap between the amount of available protein sequence due to the development of next generation sequencing technology (NGS) and slow and expensive experimental extraction of useful information like annotation of protein sequence in different functional aspects, is ever widening, which can be reduced by employing automatic function prediction (AFP) approaches. Gene Ontology (GO), comprising of more than 40, 000 classes, defines three aspects of protein function names Biological Process (BP), Cellular Component (CC), Molecular Function (MF). Multiple functions of a single protein, has made automatic function prediction a large-scale, multi-class, multi-label task. In this paper, we present DEEPGONET, a novel cascaded convolutional and recurrent neural network, to predict the top-level hierarchy of GO ontology. The network takes the primary sequence of protein as input which makes it more useful than other prevailing state-of-the-art deep learning based methods with multi-modal input, making them less applicable for proteins where only primary sequence is available. All predictions of different protein functions of our network are performed by the same architecture, a proof of better generalization as demonstrated by promising performance on a variety of organisms while trained on Homo sapiens only, which is made possible by efficient exploration of vast output space by leveraging hierarchical relationship among GO classes. The promising performance of our model makes it a potential avenue for directing experimental protein functions exploration efficiently by vastly eliminating possible routes which is done by the exploring only the suggested routes from our model. Our proposed model is also very simple and efficient in terms of computational time and space compared to other architectures in literature.