 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEfficient Approximations of Complete Interatomic Potentials for Crystal Property Prediction
Paper and Code
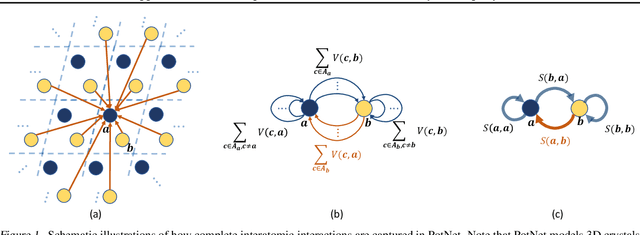
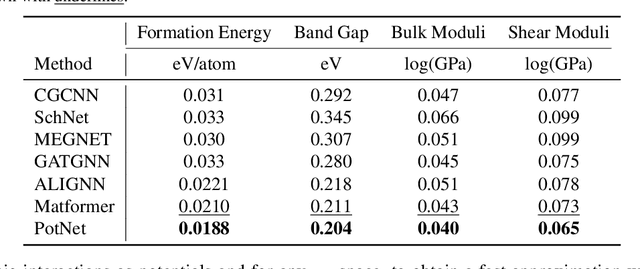
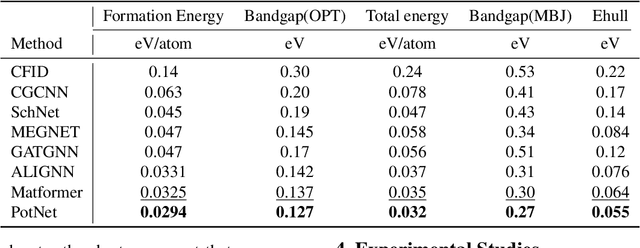
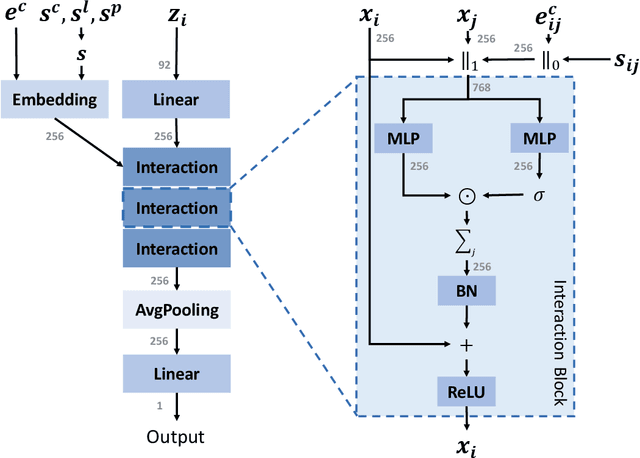
We study property prediction for crystal materials. A crystal structure consists of a minimal unit cell that is repeated infinitely in 3D space. How to accurately represent such repetitive structures in machine learning models remains unresolved. Current methods construct graphs by establishing edges only between nearby nodes, thereby failing to faithfully capture infinite repeating patterns and distant interatomic interactions. In this work, we propose several innovations to overcome these limitations. First, we propose to model physics-principled interatomic potentials directly instead of only using distances as in many existing methods. These potentials include the Coulomb potential, London dispersion potential, and Pauli repulsion potential. Second, we model the complete set of potentials among all atoms, instead of only between nearby atoms as in existing methods. This is enabled by our approximations of infinite potential summations with provable error bounds. We further develop efficient algorithms to compute the approximations. Finally, we propose to incorporate our computations of complete interatomic potentials into message passing neural networks for representation learning. We perform experiments on the JARVIS and Materials Project benchmarks for evaluation. Results show that the use of interatomic potentials and complete interatomic potentials leads to consistent performance improvements with reasonable computational costs. Our code is publicly available as part of the AIRS library (https://github.com/divelab/AIRS/tree/main/OpenMat/PotNet).