 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeColmena: Scalable Machine-Learning-Based Steering of Ensemble Simulations for High Performance Computing
Oct 06, 2021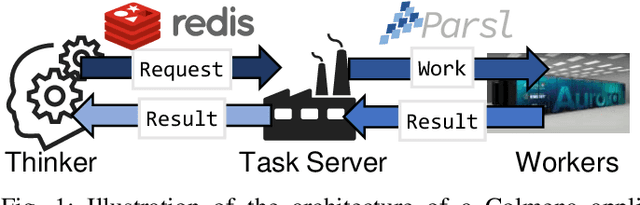
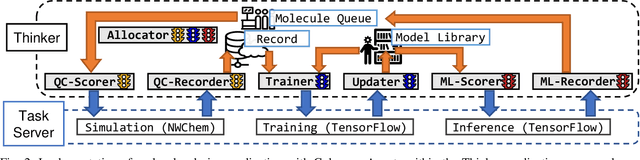
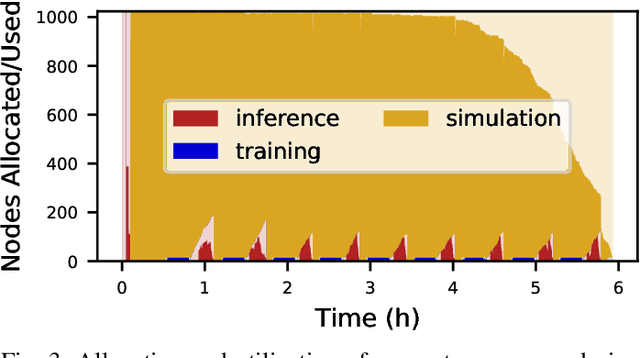
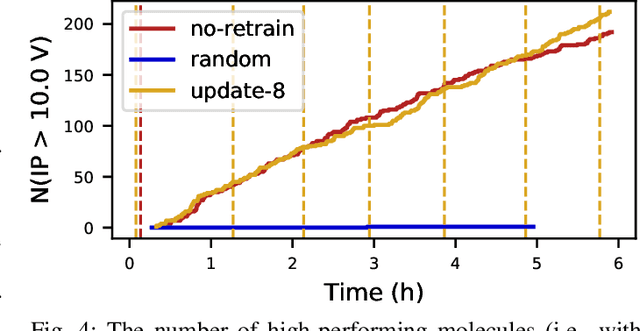
Scientific applications that involve simulation ensembles can be accelerated greatly by using experiment design methods to select the best simulations to perform. Methods that use machine learning (ML) to create proxy models of simulations show particular promise for guiding ensembles but are challenging to deploy because of the need to coordinate dynamic mixes of simulation and learning tasks. We present Colmena, an open-source Python framework that allows users to steer campaigns by providing just the implementations of individual tasks plus the logic used to choose which tasks to execute when. Colmena handles task dispatch, results collation, ML model invocation, and ML model (re)training, using Parsl to execute tasks on HPC systems. We describe the design of Colmena and illustrate its capabilities by applying it to electrolyte design, where it both scales to 65536 CPUs and accelerates the discovery rate for high-performance molecules by a factor of 100 over unguided searches.
Machine Learning Prediction of Accurate Atomization Energies of Organic Molecules from Low-Fidelity Quantum Chemical Calculations
Jun 07, 2019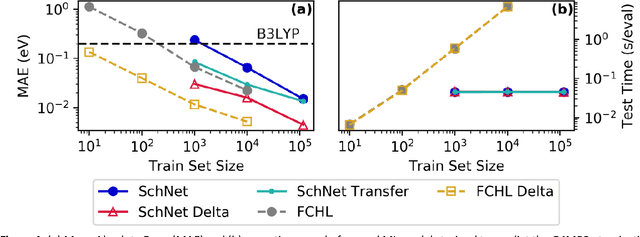
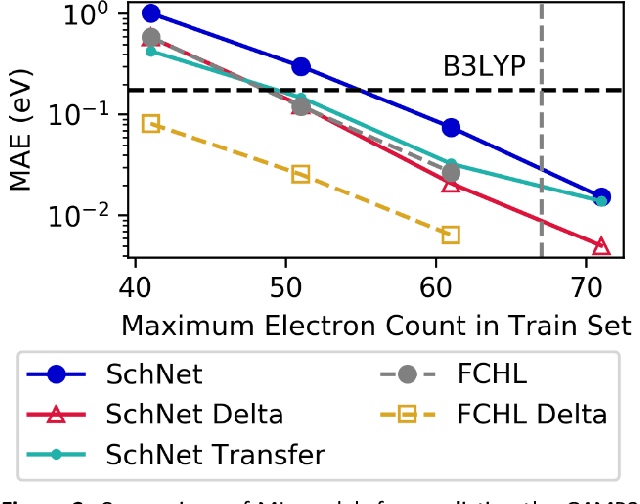
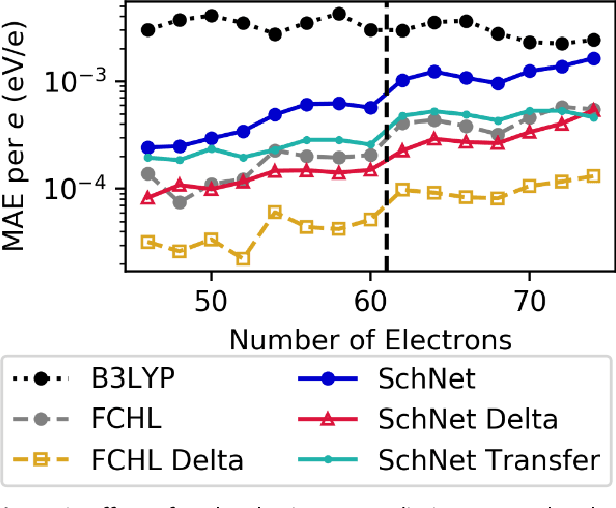
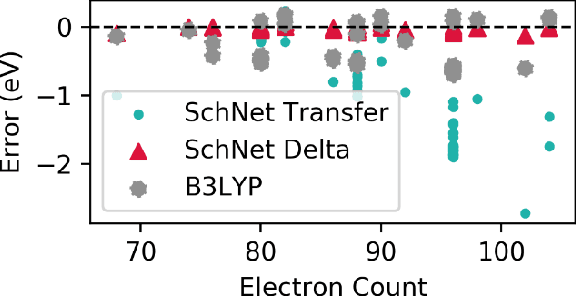
Recent studies illustrate how machine learning (ML) can be used to bypass a core challenge of molecular modeling: the tradeoff between accuracy and computational cost. Here, we assess multiple ML approaches for predicting the atomization energy of organic molecules. Our resulting models learn the difference between low-fidelity, B3LYP, and high-accuracy, G4MP2, atomization energies, and predict the G4MP2 atomization energy to 0.005 eV (mean absolute error) for molecules with less than 9 heavy atoms and 0.012 eV for a small set of molecules with between 10 and 14 heavy atoms. Our two best models, which have different accuracy/speed tradeoffs, enable the efficient prediction of G4MP2-level energies for large molecules and are available through a simple web interface.