 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMachine Learning Prediction of Accurate Atomization Energies of Organic Molecules from Low-Fidelity Quantum Chemical Calculations
Paper and Code
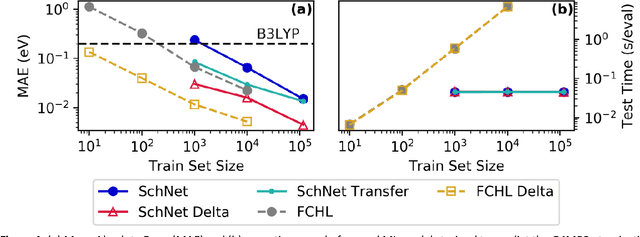
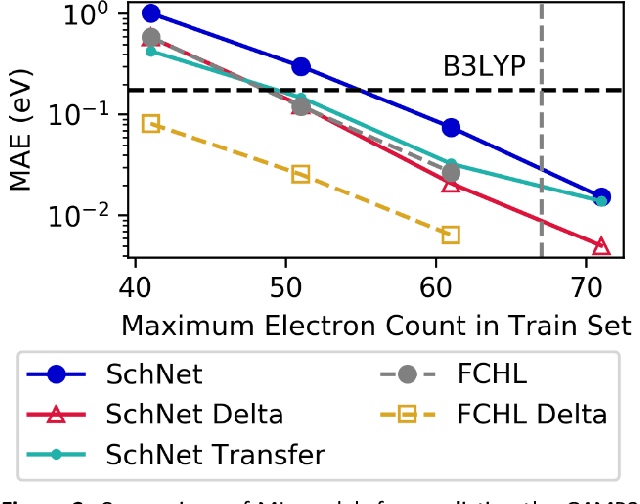
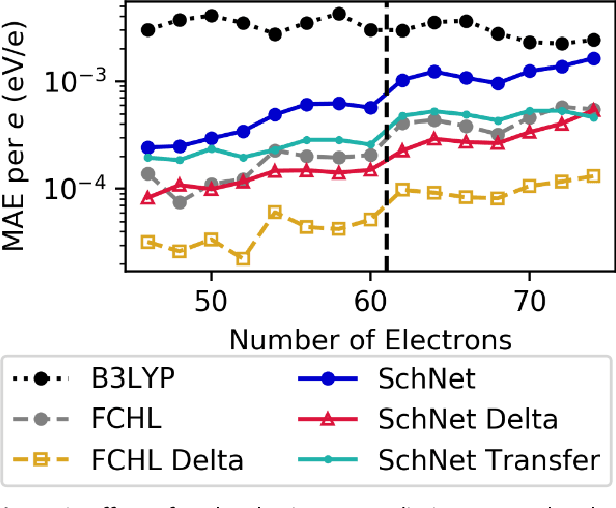
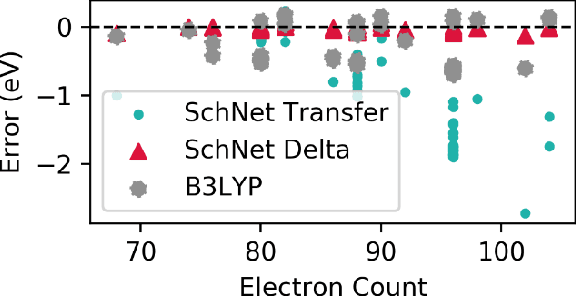
Recent studies illustrate how machine learning (ML) can be used to bypass a core challenge of molecular modeling: the tradeoff between accuracy and computational cost. Here, we assess multiple ML approaches for predicting the atomization energy of organic molecules. Our resulting models learn the difference between low-fidelity, B3LYP, and high-accuracy, G4MP2, atomization energies, and predict the G4MP2 atomization energy to 0.005 eV (mean absolute error) for molecules with less than 9 heavy atoms and 0.012 eV for a small set of molecules with between 10 and 14 heavy atoms. Our two best models, which have different accuracy/speed tradeoffs, enable the efficient prediction of G4MP2-level energies for large molecules and are available through a simple web interface.