 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHiPerRAG: High-Performance Retrieval Augmented Generation for Scientific Insights
May 07, 2025The volume of scientific literature is growing exponentially, leading to underutilized discoveries, duplicated efforts, and limited cross-disciplinary collaboration. Retrieval Augmented Generation (RAG) offers a way to assist scientists by improving the factuality of Large Language Models (LLMs) in processing this influx of information. However, scaling RAG to handle millions of articles introduces significant challenges, including the high computational costs associated with parsing documents and embedding scientific knowledge, as well as the algorithmic complexity of aligning these representations with the nuanced semantics of scientific content. To address these issues, we introduce HiPerRAG, a RAG workflow powered by high performance computing (HPC) to index and retrieve knowledge from more than 3.6 million scientific articles. At its core are Oreo, a high-throughput model for multimodal document parsing, and ColTrast, a query-aware encoder fine-tuning algorithm that enhances retrieval accuracy by using contrastive learning and late-interaction techniques. HiPerRAG delivers robust performance on existing scientific question answering benchmarks and two new benchmarks introduced in this work, achieving 90% accuracy on SciQ and 76% on PubMedQA-outperforming both domain-specific models like PubMedGPT and commercial LLMs such as GPT-4. Scaling to thousands of GPUs on the Polaris, Sunspot, and Frontier supercomputers, HiPerRAG delivers million document-scale RAG workflows for unifying scientific knowledge and fostering interdisciplinary innovation.
AdaParse: An Adaptive Parallel PDF Parsing and Resource Scaling Engine
Apr 23, 2025Language models for scientific tasks are trained on text from scientific publications, most distributed as PDFs that require parsing. PDF parsing approaches range from inexpensive heuristics (for simple documents) to computationally intensive ML-driven systems (for complex or degraded ones). The choice of the "best" parser for a particular document depends on its computational cost and the accuracy of its output. To address these issues, we introduce an Adaptive Parallel PDF Parsing and Resource Scaling Engine (AdaParse), a data-driven strategy for assigning an appropriate parser to each document. We enlist scientists to select preferred parser outputs and incorporate this information through direct preference optimization (DPO) into AdaParse, thereby aligning its selection process with human judgment. AdaParse then incorporates hardware requirements and predicted accuracy of each parser to orchestrate computational resources efficiently for large-scale parsing campaigns. We demonstrate that AdaParse, when compared to state-of-the-art parsers, improves throughput by $17\times$ while still achieving comparable accuracy (0.2 percent better) on a benchmark set of 1000 scientific documents. AdaParse's combination of high accuracy and parallel scalability makes it feasible to parse large-scale scientific document corpora to support the development of high-quality, trillion-token-scale text datasets. The implementation is available at https://github.com/7shoe/AdaParse/
BioNeMo Framework: a modular, high-performance library for AI model development in drug discovery
Nov 15, 2024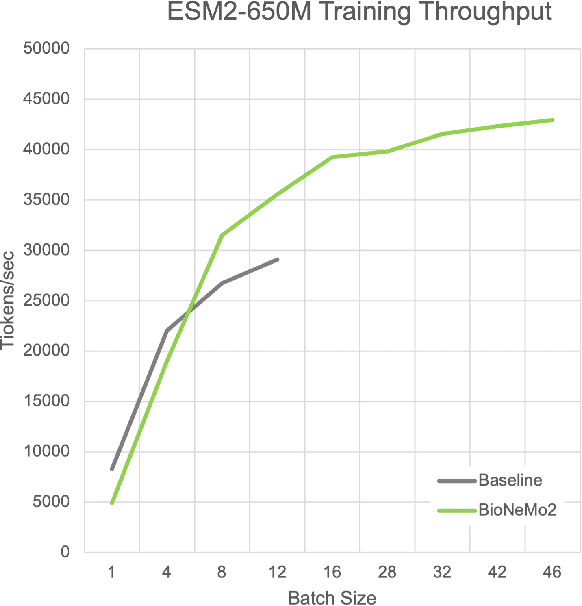
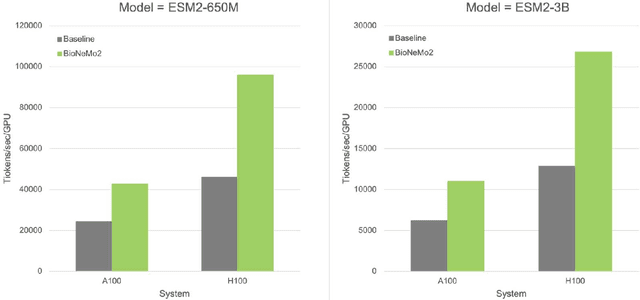
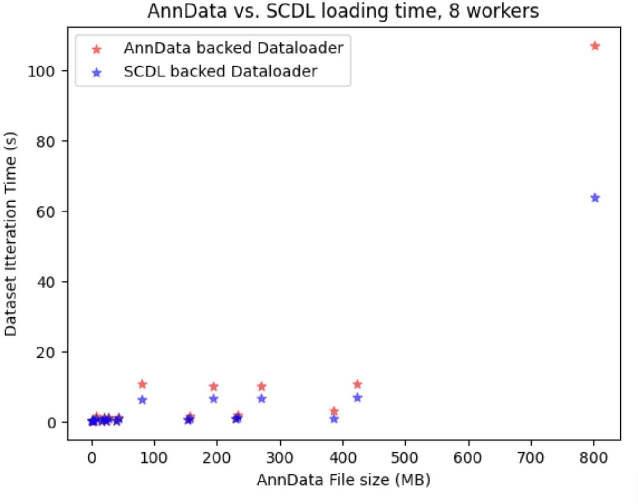
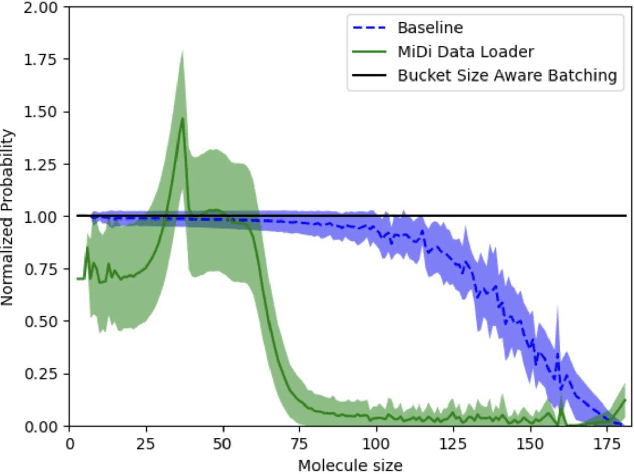
Artificial Intelligence models encoding biology and chemistry are opening new routes to high-throughput and high-quality in-silico drug development. However, their training increasingly relies on computational scale, with recent protein language models (pLM) training on hundreds of graphical processing units (GPUs). We introduce the BioNeMo Framework to facilitate the training of computational biology and chemistry AI models across hundreds of GPUs. Its modular design allows the integration of individual components, such as data loaders, into existing workflows and is open to community contributions. We detail technical features of the BioNeMo Framework through use cases such as pLM pre-training and fine-tuning. On 256 NVIDIA A100s, BioNeMo Framework trains a three billion parameter BERT-based pLM on over one trillion tokens in 4.2 days. The BioNeMo Framework is open-source and free for everyone to use.
LSHBloom: Memory-efficient, Extreme-scale Document Deduplication
Nov 06, 2024Deduplication is a major focus for assembling and curating training datasets for large language models (LLM) -- detecting and eliminating additional instances of the same content -- in large collections of technical documents. Unrestrained, duplicates in the training dataset increase training costs and lead to undesirable properties such as memorization in trained models or cheating on evaluation. Contemporary approaches to document-level deduplication are often extremely expensive in both runtime and memory. We propose LSHBloom, an extension to MinhashLSH, which replaces the expensive LSHIndex with lightweight Bloom filters. LSHBloom demonstrates the same deduplication performance as MinhashLSH with only a marginal increase in false positives (as low as 1e-5 in our experiments); demonstrates competitive runtime (270\% faster than MinhashLSH on peS2o); and, crucially, uses just 0.6\% of the disk space required by MinhashLSH to deduplicate peS2o. We demonstrate that this space advantage scales with increased dataset size -- at the extreme scale of several billion documents, LSHBloom promises a 250\% speedup and a 54$\times$ space advantage over traditional MinHashLSH scaling deduplication of text datasets to many billions of documents.
DeepSpeed4Science Initiative: Enabling Large-Scale Scientific Discovery through Sophisticated AI System Technologies
Oct 11, 2023



In the upcoming decade, deep learning may revolutionize the natural sciences, enhancing our capacity to model and predict natural occurrences. This could herald a new era of scientific exploration, bringing significant advancements across sectors from drug development to renewable energy. To answer this call, we present DeepSpeed4Science initiative (deepspeed4science.ai) which aims to build unique capabilities through AI system technology innovations to help domain experts to unlock today's biggest science mysteries. By leveraging DeepSpeed's current technology pillars (training, inference and compression) as base technology enablers, DeepSpeed4Science will create a new set of AI system technologies tailored for accelerating scientific discoveries by addressing their unique complexity beyond the common technical approaches used for accelerating generic large language models (LLMs). In this paper, we showcase the early progress we made with DeepSpeed4Science in addressing two of the critical system challenges in structural biology research.
Exploring Benchmarks for Self-Driving Labs using Color Matching
Sep 30, 2023Self Driving Labs (SDLs) that combine automation of experimental procedures with autonomous decision making are gaining popularity as a means of increasing the throughput of scientific workflows. The task of identifying quantities of supplied colored pigments that match a target color, the color matching problem, provides a simple and flexible SDL test case, as it requires experiment proposal, sample creation, and sample analysis, three common components in autonomous discovery applications. We present a robotic solution to the color matching problem that allows for fully autonomous execution of a color matching protocol. Our solution leverages the WEI science factory platform to enable portability across different robotic hardware, the use of alternative optimization methods for continuous refinement, and automated publication of results for experiment tracking and post-hoc analysis.
Towards a Modular Architecture for Science Factories
Aug 18, 2023Advances in robotic automation, high-performance computing (HPC), and artificial intelligence (AI) encourage us to conceive of science factories: large, general-purpose computation- and AI-enabled self-driving laboratories (SDLs) with the generality and scale needed both to tackle large discovery problems and to support thousands of scientists. Science factories require modular hardware and software that can be replicated for scale and (re)configured to support many applications. To this end, we propose a prototype modular science factory architecture in which reconfigurable modules encapsulating scientific instruments are linked with manipulators to form workcells, that can themselves be combined to form larger assemblages, and linked with distributed computing for simulation, AI model training and inference, and related tasks. Workflows that perform sets of actions on modules can be specified, and various applications, comprising workflows plus associated computational and data manipulation steps, can be run concurrently. We report on our experiences prototyping this architecture and applying it in experiments involving 15 different robotic apparatus, five applications (one in education, two in biology, two in materials), and a variety of workflows, across four laboratories. We describe the reuse of modules, workcells, and workflows in different applications, the migration of applications between workcells, and the use of digital twins, and suggest directions for future work aimed at yet more generality and scalability. Code and data are available at https://ad-sdl.github.io/wei2023 and in the Supplementary Information
Artificial Intelligence Advances for De Novo Molecular Structure Modeling in Cryo-EM
Feb 24, 2021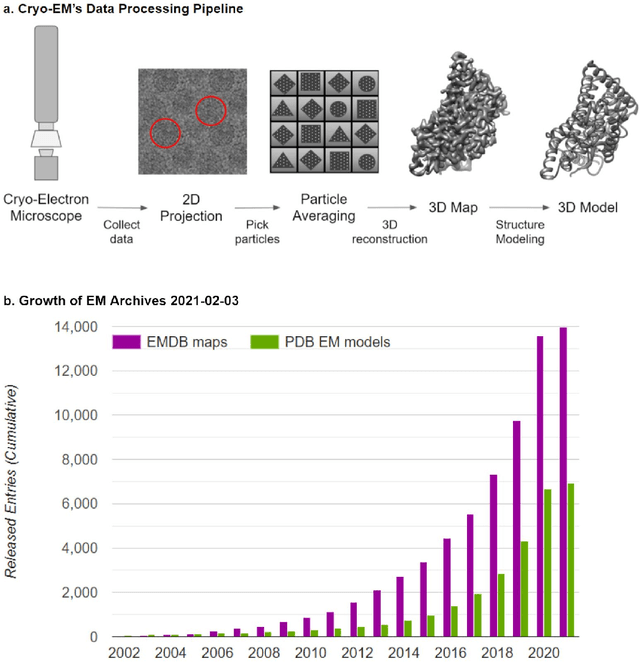
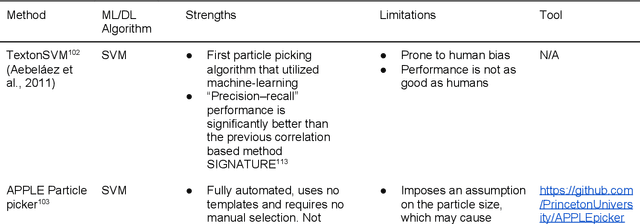


Cryo-electron microscopy (cryo-EM) has become a major experimental technique to determine the structures of large protein complexes and molecular assemblies, as evidenced by the 2017 Nobel Prize. Although cryo-EM has been drastically improved to generate high-resolution three-dimensional (3D) maps that contain detailed structural information about macromolecules, the computational methods for using the data to automatically build structure models are lagging far behind. The traditional cryo-EM model building approach is template-based homology modeling. Manual de novo modeling is very time-consuming when no template model is found in the database. In recent years, de novo cryo-EM modeling using machine learning (ML) and deep learning (DL) has ranked among the top-performing methods in macromolecular structure modeling. Deep-learning-based de novo cryo-EM modeling is an important application of artificial intelligence, with impressive results and great potential for the next generation of molecular biomedicine. Accordingly, we systematically review the representative ML/DL-based de novo cryo-EM modeling methods. And their significances are discussed from both practical and methodological viewpoints. We also briefly describe the background of cryo-EM data processing workflow. Overall, this review provides an introductory guide to modern research on artificial intelligence (AI) for de novo molecular structure modeling and future directions in this emerging field.