 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBeyond Current Boundaries: Integrating Deep Learning and AlphaFold for Enhanced Protein Structure Prediction from Low-Resolution Cryo-EM Maps
Oct 30, 2024


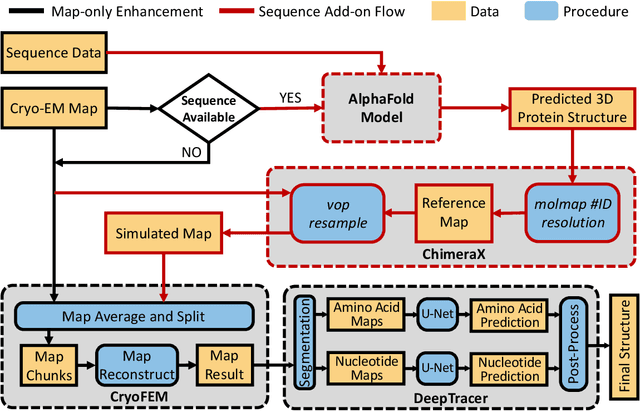
Constructing atomic models from cryo-electron microscopy (cryo-EM) maps is a crucial yet intricate task in structural biology. While advancements in deep learning, such as convolutional neural networks (CNNs) and graph neural networks (GNNs), have spurred the development of sophisticated map-to-model tools like DeepTracer and ModelAngelo, their efficacy notably diminishes with low-resolution maps beyond 4 {\AA}. To address this shortfall, our research introduces DeepTracer-LowResEnhance, an innovative framework that synergizes a deep learning-enhanced map refinement technique with the power of AlphaFold. This methodology is designed to markedly improve the construction of models from low-resolution cryo-EM maps. DeepTracer-LowResEnhance was rigorously tested on a set of 37 protein cryo-EM maps, with resolutions ranging between 2.5 to 8.4 {\AA}, including 22 maps with resolutions lower than 4 {\AA}. The outcomes were compelling, demonstrating that 95.5\% of the low-resolution maps exhibited a significant uptick in the count of total predicted residues. This denotes a pronounced improvement in atomic model building for low-resolution maps. Additionally, a comparative analysis alongside Phenix's auto-sharpening functionality delineates DeepTracer-LowResEnhance's superior capability in rendering more detailed and precise atomic models, thereby pushing the boundaries of current computational structural biology methodologies.
Self-Attention Generative Adversarial Network for Iterative Reconstruction of CT Images
Dec 23, 2021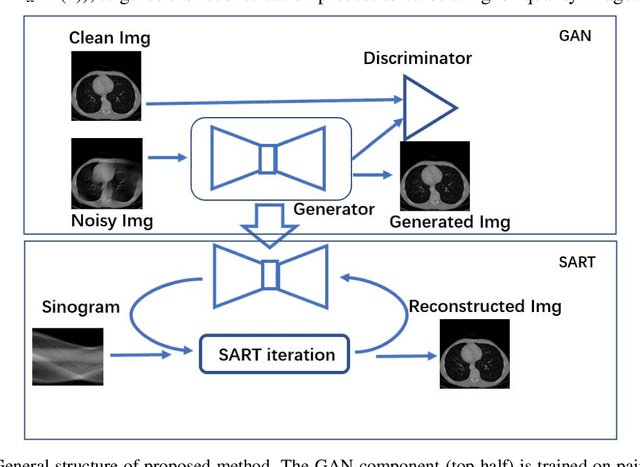
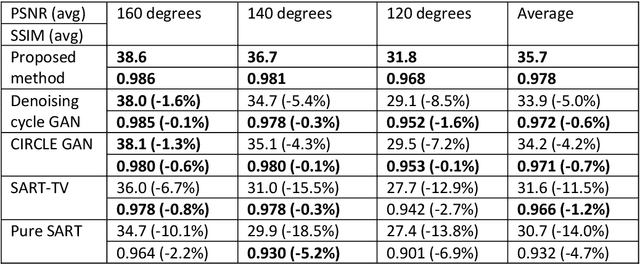
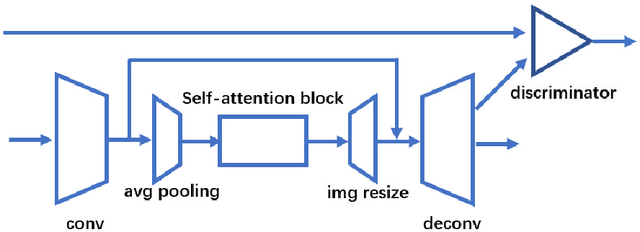
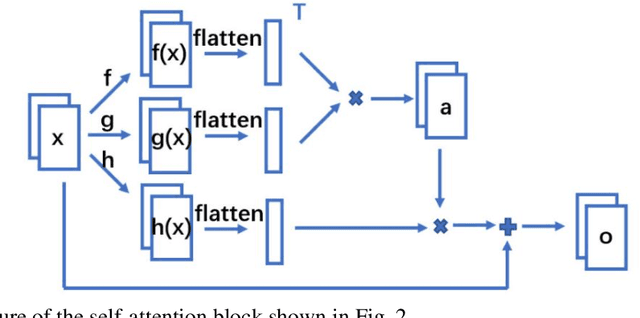
Computed tomography (CT) uses X-ray measurements taken from sensors around the body to generate tomographic images of the human body. Conventional reconstruction algorithms can be used if the X-ray data are adequately sampled and of high quality; however, concerns such as reducing dose to the patient, or geometric limitations on data acquisition, may result in low quality or incomplete data. Images reconstructed from these data using conventional methods are of poor quality, due to noise and other artifacts. The aim of this study is to train a single neural network to reconstruct high-quality CT images from noisy or incomplete CT scan data, including low-dose, sparse-view, and limited-angle scenarios. To accomplish this task, we train a generative adversarial network (GAN) as a signal prior, to be used in conjunction with the iterative simultaneous algebraic reconstruction technique (SART) for CT data. The network includes a self-attention block to model long-range dependencies in the data. We compare our Self-Attention GAN for CT image reconstruction with several state-of-the-art approaches, including denoising cycle GAN, CIRCLE GAN, and a total variation superiorized algorithm. Our approach is shown to have comparable overall performance to CIRCLE GAN, while outperforming the other two approaches.
Artificial Intelligence Advances for De Novo Molecular Structure Modeling in Cryo-EM
Feb 24, 2021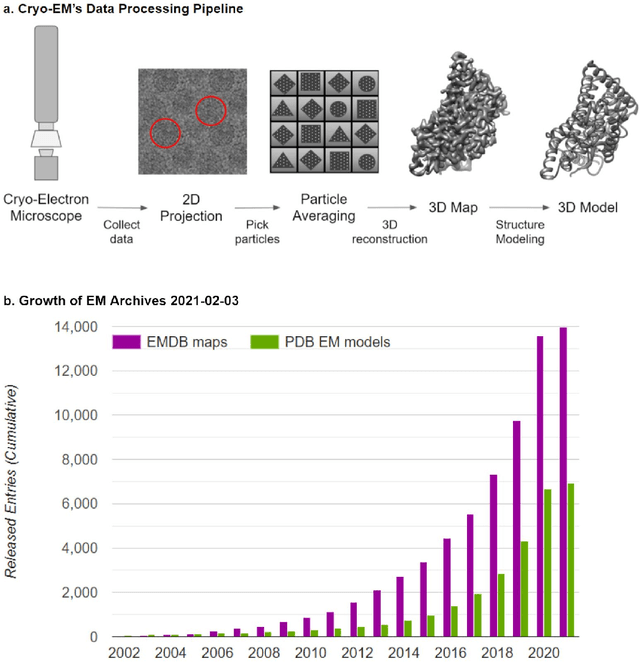
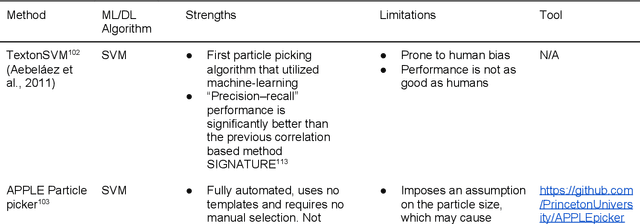


Cryo-electron microscopy (cryo-EM) has become a major experimental technique to determine the structures of large protein complexes and molecular assemblies, as evidenced by the 2017 Nobel Prize. Although cryo-EM has been drastically improved to generate high-resolution three-dimensional (3D) maps that contain detailed structural information about macromolecules, the computational methods for using the data to automatically build structure models are lagging far behind. The traditional cryo-EM model building approach is template-based homology modeling. Manual de novo modeling is very time-consuming when no template model is found in the database. In recent years, de novo cryo-EM modeling using machine learning (ML) and deep learning (DL) has ranked among the top-performing methods in macromolecular structure modeling. Deep-learning-based de novo cryo-EM modeling is an important application of artificial intelligence, with impressive results and great potential for the next generation of molecular biomedicine. Accordingly, we systematically review the representative ML/DL-based de novo cryo-EM modeling methods. And their significances are discussed from both practical and methodological viewpoints. We also briefly describe the background of cryo-EM data processing workflow. Overall, this review provides an introductory guide to modern research on artificial intelligence (AI) for de novo molecular structure modeling and future directions in this emerging field.
A Raspberry Pi-based Traumatic Brain Injury Detection System for Single-Channel Electroencephalogram
Jan 29, 2021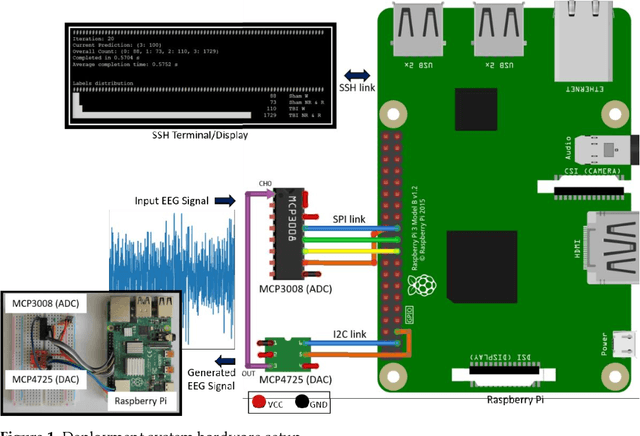
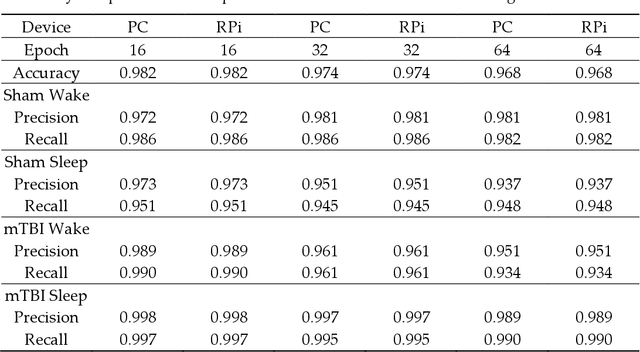
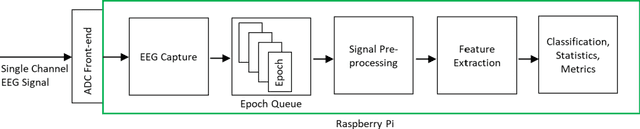
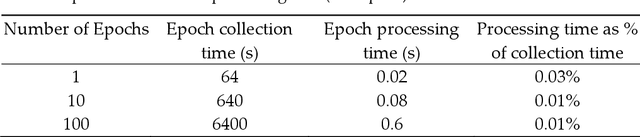
Traumatic Brain Injury (TBI) is a common cause of death and disability. However, existing tools for TBI diagnosis are either subjective or require extensive clinical setup and expertise. The increasing affordability and reduction in size of relatively high-performance computing systems combined with promising results from TBI related machine learning research make it possible to create compact and portable systems for early detection of TBI. This work describes a Raspberry Pi based portable, real-time data acquisition, and automated processing system that uses machine learning to efficiently identify TBI and automatically score sleep stages from a single-channel Electroen-cephalogram (EEG) signal. We discuss the design, implementation, and verification of the system that can digitize EEG signal using an Analog to Digital Converter (ADC) and perform real-time signal classification to detect the presence of mild TBI (mTBI). We utilize Convolutional Neural Networks (CNN) and XGBoost based predictive models to evaluate the performance and demonstrate the versatility of the system to operate with multiple types of predictive models. We achieve a peak classification accuracy of more than 90% with a classification time of less than 1 s across 16 s - 64 s epochs for TBI vs control conditions. This work can enable development of systems suitable for field use without requiring specialized medical equipment for early TBI detection applications and TBI research. Further, this work opens avenues to implement connected, real-time TBI related health and wellness monitoring systems.