 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThermodynamic assessment of machine learning models for solid-state synthesis prediction
Feb 03, 2026Machine learning models have recently emerged to predict whether hypothetical solid-state materials can be synthesized. These models aim to circumvent direct first-principles modeling of solid-state phase transformations, instead learning from large databases of successfully synthesized materials. Here, we assess the alignment of several recently introduced synthesis prediction models with material and reaction thermodynamics, quantified by the energy with respect to the convex hull and a metric accounting for thermodynamic selectivity of enumerated synthesis reactions. A dataset of successful synthesis recipes was used to determine the likely bounds on both quantities beyond which materials can be deemed unlikely to be synthesized. With these bounds as context, thermodynamic quantities were computed using the CHGNet foundation potential for thousands of new hypothetical materials generated using the Chemeleon generative model. Four recently published machine learning models for synthesizability prediction were applied to this same dataset, and the resultant predictions were considered against computed thermodynamics. We find these models generally overpredict the likelihood of synthesis, but some model scores do trend with thermodynamic heuristics, assigning lower scores to materials that are less stable or do not have an available synthesis recipe that is calculated to be thermodynamically selective. In total, this work identifies existing gaps in machine learning models for materials synthesis and introduces a new approach to assess their quality in the absence of extensive negative examples (failed syntheses).
Establishing baselines for generative discovery of inorganic crystals
Jan 04, 2025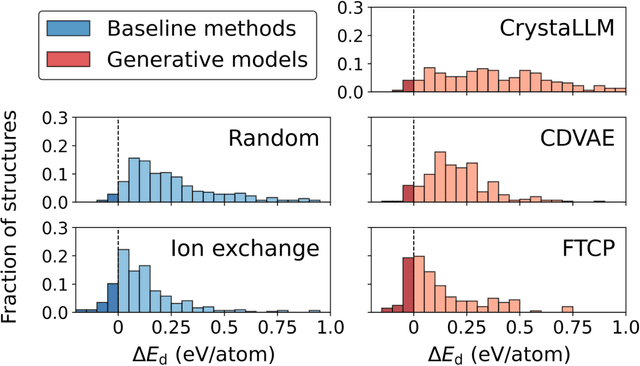
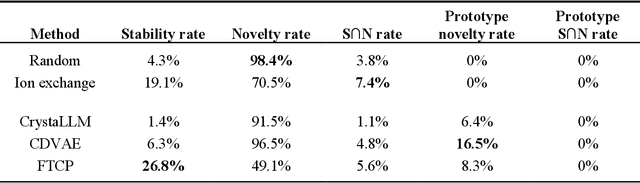
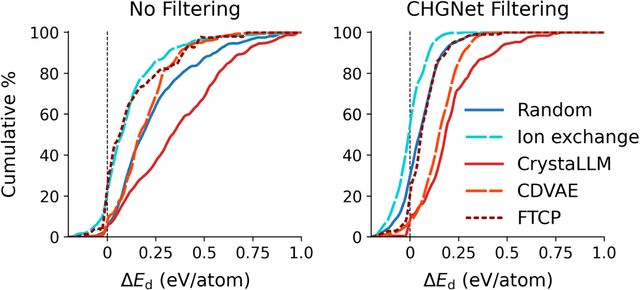
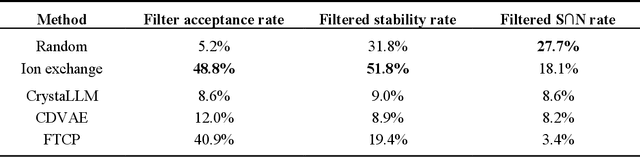
Generative artificial intelligence offers a promising avenue for materials discovery, yet its advantages over traditional methods remain unclear. In this work, we introduce and benchmark two baseline approaches - random enumeration of charge-balanced prototypes and data-driven ion exchange of known compounds - against three generative models: a variational autoencoder, a large language model, and a diffusion model. Our results show that established methods such as ion exchange perform comparably well in generating stable materials, although many of these materials tend to closely resemble known compounds. In contrast, generative models excel at proposing novel structural frameworks and, when sufficient training data is available, can more effectively target properties such as electronic band gap and bulk modulus while maintaining a high stability rate. To enhance the performance of both the baseline and generative approaches, we implement a post-generation screening step in which all proposed structures are passed through stability and property filters from pre-trained machine learning models including universal interatomic potentials. This low-cost filtering step leads to substantial improvement in the success rates of all methods, remains computationally efficient, and ultimately provides a practical pathway toward more effective generative strategies for materials discovery.
Accelerating the prediction of inorganic surfaces with machine learning interatomic potentials
Dec 18, 2023The surface properties of solid-state materials often dictate their functionality, especially for applications where nanoscale effects become important. The relevant surface(s) and their properties are determined, in large part, by the materials synthesis or operating conditions. These conditions dictate thermodynamic driving forces and kinetic rates responsible for yielding the observed surface structure and morphology. Computational surface science methods have long been applied to connect thermochemical conditions to surface phase stability, particularly in the heterogeneous catalysis and thin film growth communities. This review provides a brief introduction to first-principles approaches to compute surface phase diagrams before introducing emerging data-driven approaches. The remainder of the review focuses on the application of machine learning, predominantly in the form of learned interatomic potentials, to study complex surfaces. As machine learning algorithms and large datasets on which to train them become more commonplace in materials science, computational methods are poised to become even more predictive and powerful for modeling the complexities of inorganic surfaces at the nanoscale.
CHGNet: Pretrained universal neural network potential for charge-informed atomistic modeling
Feb 28, 2023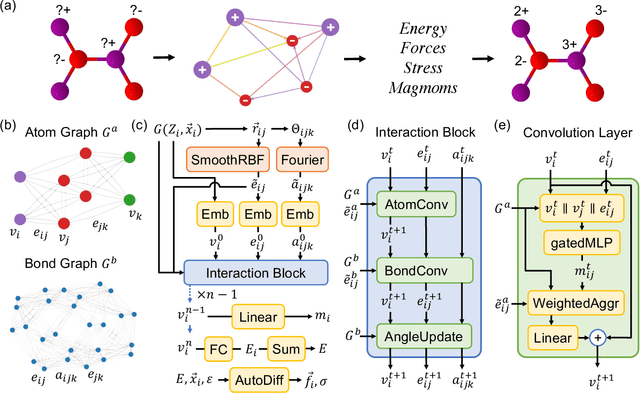
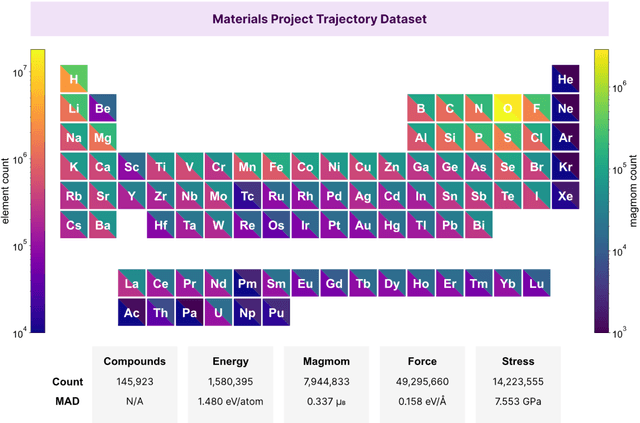
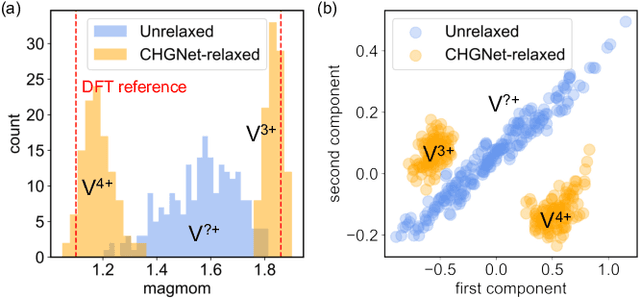
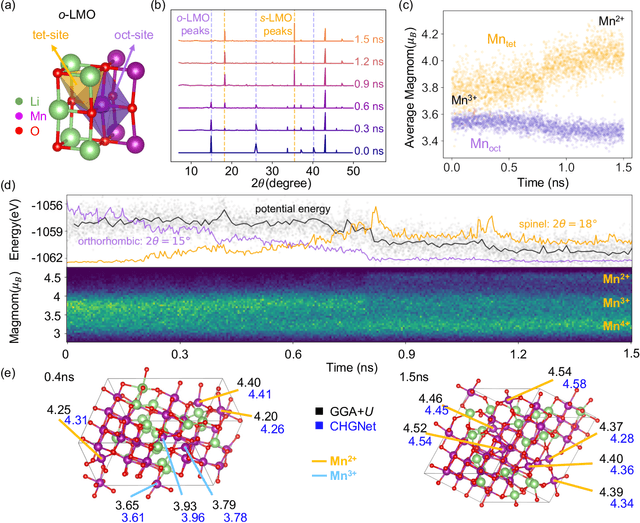
The simulation of large-scale systems with complex electron interactions remains one of the greatest challenges for the atomistic modeling of materials. Although classical force-fields often fail to describe the coupling between electronic states and ionic rearrangements, the more accurate \textit{ab-initio} molecular dynamics suffers from computational complexity that prevents long-time and large-scale simulations, which are essential to study many technologically relevant phenomena, such as reactions, ion migrations, phase transformations, and degradation. In this work, we present the Crystal Hamiltonian Graph neural Network (CHGNet) as a novel machine-learning interatomic potential (MLIP), using a graph-neural-network-based force-field to model a universal potential energy surface. CHGNet is pretrained on the energies, forces, stresses, and magnetic moments from the Materials Project Trajectory Dataset, which consists of over 10 years of density functional theory static and relaxation trajectories of $\sim 1.5$ million inorganic structures. The explicit inclusion of magnetic moments enables CHGNet to learn and accurately represent the orbital occupancy of electrons, enhancing its capability to describe both atomic and electronic degrees of freedom. We demonstrate several applications of CHGNet in solid-state materials, including charge-informed molecular dynamics in Li$_x$MnO$_2$, the finite temperature phase diagram for Li$_x$FePO$_4$ and Li diffusion in garnet conductors. We critically analyze the significance of including charge information for capturing appropriate chemistry, and we provide new insights into ionic systems with additional electronic degrees of freedom that can not be observed by previous MLIPs.
Inorganic synthesis recommendation by machine learning materials similarity from scientific literature
Feb 05, 2023Synthesis prediction is a key accelerator for the rapid design of advanced materials. However, determining synthesis variables such as the choice of precursor materials, operations, and conditions is challenging for inorganic materials because the sequence of reactions during heating is not well understood. In this work, we use a knowledge base of 29,900 solid-state synthesis recipes, text-mined from the scientific literature, to automatically learn which precursors to recommend for the synthesis of a novel target material. The data-driven approach learns chemical similarity of materials and refers the synthesis of a new target to precedent synthesis procedures of similar materials, mimicking human synthesis design. When proposing five precursor sets for each of 2,654 unseen test target materials, the recommendation strategy achieves a success rate of at least 82%. Our approach captures decades of heuristic synthesis data in a mathematical form, making it accessible for use in recommendation engines and autonomous laboratories.
A probabilistic deep learning approach to automate the interpretation of multi-phase diffraction spectra
Mar 30, 2021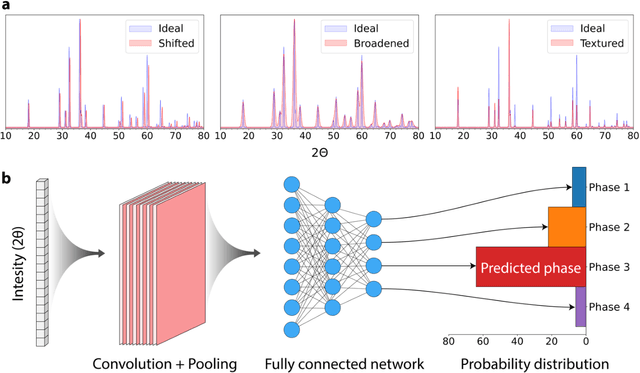
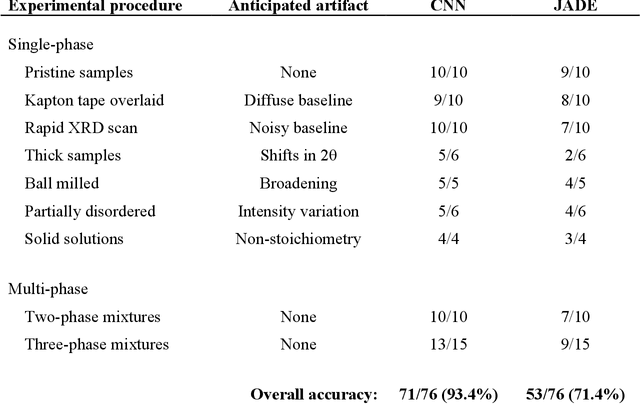
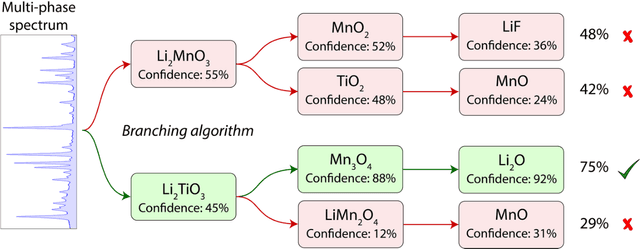
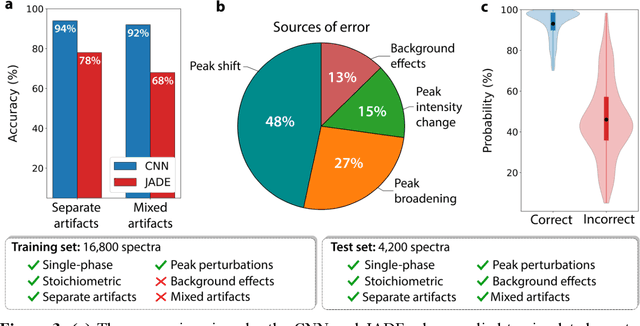
Autonomous synthesis and characterization of inorganic materials requires the automatic and accurate analysis of X-ray diffraction spectra. For this task, we designed a probabilistic deep learning algorithm to identify complex multi-phase mixtures. At the core of this algorithm lies an ensemble convolutional neural network trained on simulated diffraction spectra, which are systematically augmented with physics-informed perturbations to account for artifacts that can arise during experimental sample preparation and synthesis. Larger perturbations associated with off-stoichiometry are also captured by supplementing the training set with hypothetical solid solutions. Spectra containing mixtures of materials are analyzed with a newly developed branching algorithm that utilizes the probabilistic nature of the neural network to explore suspected mixtures and identify the set of phases that maximize confidence in the prediction. Our model is benchmarked on simulated and experimentally measured diffraction spectra, showing exceptional performance with accuracies exceeding those given by previously reported methods based on profile matching and deep learning. We envision that the algorithm presented here may be integrated in experimental workflows to facilitate the high-throughput and autonomous discovery of inorganic materials.