 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHessian-informed machine learning interatomic potential towards bridging theory and experiments
Mar 26, 2026Local curvature of potential energy surfaces is critical for predicting certain experimental observables of molecules and materials from first principles, yet it remains far beyond reach for complex systems. In this work, we introduce a Hessian-informed Machine Learning Interatomic Potential (Hi-MLIP) that captures such curvature reliably, thereby enabling accurate analysis of associated thermodynamic and kinetic phenomena. To make Hessian supervision practically viable, we develop a highly efficient training protocol, termed Hessian INformed Training (HINT), achieving two to four orders of magnitude reduction for the requirement of expensive Hessian labels. HINT integrates critical techniques, including Hessian pre-training, configuration sampling, curriculum learning and stochastic projection Hessian loss. Enabled by HINT, Hi-MLIP significantly improves transition-state search and brings Gibbs free-energy predictions close to chemical accuracy especially in data-scarce regimes. Our framework also enables accurate treatment of strongly anharmonic hydrides, reproducing phonon renormalization and superconducting critical temperatures in close agreement with experiment while bypassing the computational bottleneck of anharmonic calculations. These results establish a practical route to enhancing curvature awareness of machine learning interatomic potentials, bridging simulation and experimental observables across a wide range of systems.
AlphaNet: Scaling Up Local Frame-based Atomistic Foundation Model
Jan 13, 2025
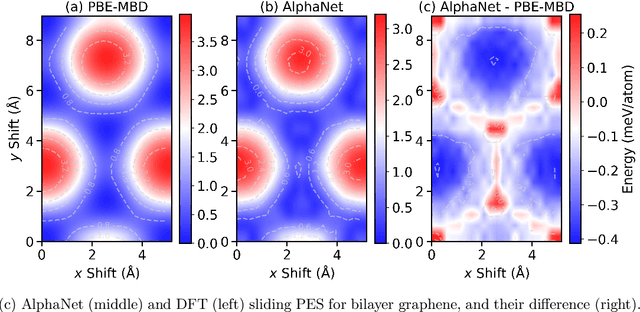
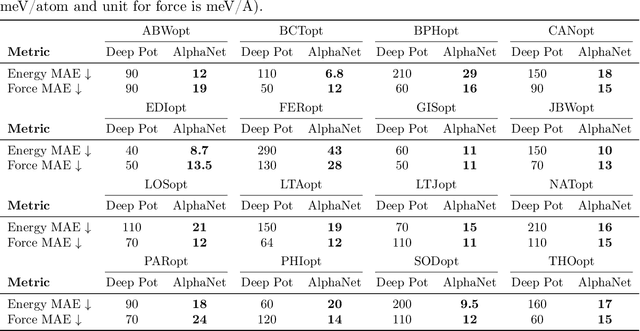
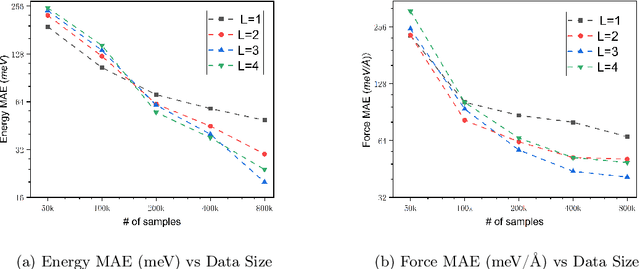
We present AlphaNet, a local frame-based equivariant model designed to achieve both accurate and efficient simulations for atomistic systems. Recently, machine learning force fields (MLFFs) have gained prominence in molecular dynamics simulations due to their advantageous efficiency-accuracy balance compared to classical force fields and quantum mechanical calculations, alongside their transferability across various systems. Despite the advancements in improving model accuracy, the efficiency and scalability of MLFFs remain significant obstacles in practical applications. AlphaNet enhances computational efficiency and accuracy by leveraging the local geometric structures of atomic environments through the construction of equivariant local frames and learnable frame transitions. We substantiate the efficacy of AlphaNet across diverse datasets, including defected graphene, formate decomposition, zeolites, and surface reactions. AlphaNet consistently surpasses well-established models, such as NequIP and DeepPot, in terms of both energy and force prediction accuracy. Notably, AlphaNet offers one of the best trade-offs between computational efficiency and accuracy among existing models. Moreover, AlphaNet exhibits scalability across a broad spectrum of system and dataset sizes, affirming its versatility.
EL-MLFFs: Ensemble Learning of Machine Leaning Force Fields
Mar 26, 2024



Machine learning force fields (MLFFs) have emerged as a promising approach to bridge the accuracy of quantum mechanical methods and the efficiency of classical force fields. However, the abundance of MLFF models and the challenge of accurately predicting atomic forces pose significant obstacles in their practical application. In this paper, we propose a novel ensemble learning framework, EL-MLFFs, which leverages the stacking method to integrate predictions from diverse MLFFs and enhance force prediction accuracy. By constructing a graph representation of molecular structures and employing a graph neural network (GNN) as the meta-model, EL-MLFFs effectively captures atomic interactions and refines force predictions. We evaluate our approach on two distinct datasets: methane molecules and methanol adsorbed on a Cu(100) surface. The results demonstrate that EL-MLFFs significantly improves force prediction accuracy compared to individual MLFFs, with the ensemble of all eight models yielding the best performance. Moreover, our ablation study highlights the crucial roles of the residual network and graph attention layers in the model's architecture. The EL-MLFFs framework offers a promising solution to the challenges of model selection and force prediction accuracy in MLFFs, paving the way for more reliable and efficient molecular simulations.
Interactive Molecular Discovery with Natural Language
Jun 21, 2023Natural language is expected to be a key medium for various human-machine interactions in the era of large language models. When it comes to the biochemistry field, a series of tasks around molecules (e.g., property prediction, molecule mining, etc.) are of great significance while having a high technical threshold. Bridging the molecule expressions in natural language and chemical language can not only hugely improve the interpretability and reduce the operation difficulty of these tasks, but also fuse the chemical knowledge scattered in complementary materials for a deeper comprehension of molecules. Based on these benefits, we propose the conversational molecular design, a novel task adopting natural language for describing and editing target molecules. To better accomplish this task, we design ChatMol, a knowledgeable and versatile generative pre-trained model, enhanced by injecting experimental property information, molecular spatial knowledge, and the associations between natural and chemical languages into it. Several typical solutions including large language models (e.g., ChatGPT) are evaluated, proving the challenge of conversational molecular design and the effectiveness of our knowledge enhancement method. Case observations and analysis are conducted to provide directions for further exploration of natural-language interaction in molecular discovery.