 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGenerative Pseudo-Force Fields for Molecular Generation
May 18, 2026Generating stable molecular conformations typically forces a tradeoff between the physical realism of energy-based relaxation and the sampling efficiency of data-driven generative models. While machine learning force fields (MLFFs) can sample stable conformations by relaxing molecular geometries according to physical forces, they require costly ab-initio training data. Conversely, diffusion models (DMs) learn from equilibrium data alone but are dependent on noise schedules and time-step conditioning. In this work, we propose generative pseudo-force fields (GPFFs) to bridge these paradigms by training an MLFF on a quadratic pseudo-potential energy surface relative to reference equilibrium structures. Because no ab-initio calculations are required for the perturbed geometries, non-equilibrium training data can be generated on the fly by perturbing the equilibria with Gaussian noise. We show that GPFFs constitute a time-step-agnostic variant of variance exploding DMs: the score comes from the predicted pseudo-forces but because force magnitudes implicitly encode the noise level, no time-step conditioning is needed. Our GPFF can hence be used as a drop-in replacement in standard diffusion sampling (ancestral, Heun) but also facilitates more efficient, adaptive variants and an MLFF inspired direct denoising scheme. Our proposed sampling algorithms support arbitrary structural priors and geometric constraints. On QM9, GPFF has 100 % validity at 256 neural function evaluations (NFE) and over 50 % at just 6 NFE, outperforming diffusion baselines across all samplers. Combined with custom priors, we showcase the fast and accurate generation process of our method in a molecular editor for a drug design setting, where a molecule is generated in real time.
Learning Hamiltonian Flow Maps: Mean Flow Consistency for Large-Timestep Molecular Dynamics
Jan 29, 2026Simulating the long-time evolution of Hamiltonian systems is limited by the small timesteps required for stable numerical integration. To overcome this constraint, we introduce a framework to learn Hamiltonian Flow Maps by predicting the mean phase-space evolution over a chosen time span $Δt$, enabling stable large-timestep updates far beyond the stability limits of classical integrators. To this end, we impose a Mean Flow consistency condition for time-averaged Hamiltonian dynamics. Unlike prior approaches, this allows training on independent phase-space samples without access to future states, avoiding expensive trajectory generation. Validated across diverse Hamiltonian systems, our method in particular improves upon molecular dynamics simulations using machine-learned force fields (MLFF). Our models maintain comparable training and inference cost, but support significantly larger integration timesteps while trained directly on widely-available trajectory-free MLFF datasets.
Doob's Lagrangian: A Sample-Efficient Variational Approach to Transition Path Sampling
Oct 10, 2024

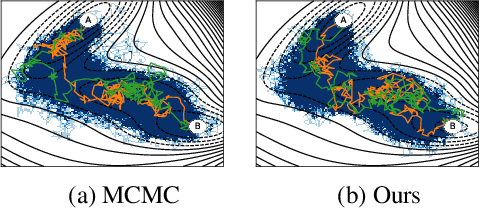
Rare event sampling in dynamical systems is a fundamental problem arising in the natural sciences, which poses significant computational challenges due to an exponentially large space of trajectories. For settings where the dynamical system of interest follows a Brownian motion with known drift, the question of conditioning the process to reach a given endpoint or desired rare event is definitively answered by Doob's h-transform. However, the naive estimation of this transform is infeasible, as it requires simulating sufficiently many forward trajectories to estimate rare event probabilities. In this work, we propose a variational formulation of Doob's $h$-transform as an optimization problem over trajectories between a given initial point and the desired ending point. To solve this optimization, we propose a simulation-free training objective with a model parameterization that imposes the desired boundary conditions by design. Our approach significantly reduces the search space over trajectories and avoids expensive trajectory simulation and inefficient importance sampling estimators which are required in existing methods. We demonstrate the ability of our method to find feasible transition paths on real-world molecular simulation and protein folding tasks.
Transition Path Sampling with Boltzmann Generator-based MCMC Moves
Dec 08, 2023



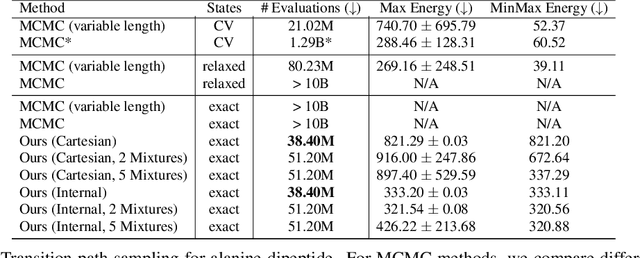
Sampling all possible transition paths between two 3D states of a molecular system has various applications ranging from catalyst design to drug discovery. Current approaches to sample transition paths use Markov chain Monte Carlo and rely on time-intensive molecular dynamics simulations to find new paths. Our approach operates in the latent space of a normalizing flow that maps from the molecule's Boltzmann distribution to a Gaussian, where we propose new paths without requiring molecular simulations. Using alanine dipeptide, we explore Metropolis-Hastings acceptance criteria in the latent space for exact sampling and investigate different latent proposal mechanisms.