 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGenerative Pseudo-Force Fields for Molecular Generation
May 18, 2026Generating stable molecular conformations typically forces a tradeoff between the physical realism of energy-based relaxation and the sampling efficiency of data-driven generative models. While machine learning force fields (MLFFs) can sample stable conformations by relaxing molecular geometries according to physical forces, they require costly ab-initio training data. Conversely, diffusion models (DMs) learn from equilibrium data alone but are dependent on noise schedules and time-step conditioning. In this work, we propose generative pseudo-force fields (GPFFs) to bridge these paradigms by training an MLFF on a quadratic pseudo-potential energy surface relative to reference equilibrium structures. Because no ab-initio calculations are required for the perturbed geometries, non-equilibrium training data can be generated on the fly by perturbing the equilibria with Gaussian noise. We show that GPFFs constitute a time-step-agnostic variant of variance exploding DMs: the score comes from the predicted pseudo-forces but because force magnitudes implicitly encode the noise level, no time-step conditioning is needed. Our GPFF can hence be used as a drop-in replacement in standard diffusion sampling (ancestral, Heun) but also facilitates more efficient, adaptive variants and an MLFF inspired direct denoising scheme. Our proposed sampling algorithms support arbitrary structural priors and geometric constraints. On QM9, GPFF has 100 % validity at 256 neural function evaluations (NFE) and over 50 % at just 6 NFE, outperforming diffusion baselines across all samplers. Combined with custom priors, we showcase the fast and accurate generation process of our method in a molecular editor for a drug design setting, where a molecule is generated in real time.
Enabling ab initio geometry optimization of strongly correlated systems with transferable deep quantum Monte Carlo
Mar 26, 2026A faithful description of chemical processes requires exploring extended regions of the molecular potential energy surface (PES), which remains challenging for strongly correlated systems. Transferable deep-learning variational Monte Carlo (VMC) offers a promising route by efficiently solving the electronic Schrödinger equation jointly across molecular geometries at consistently high accuracy, yet its stochastic nature renders direct exploration of molecular configuration space nontrivial. Here, we present a framework for highly accurate ab initio exploration of PESs that combines transferable deep-learning VMC with a cost-effective estimation of energies, forces, and Hessians. By continuously sampling nuclear configurations during VMC optimization of electronic wave functions, we obtain transferable descriptions that achieve zero-shot chemical accuracy within chemically relevant distributions of molecular geometries. Throughout the subsequent characterization of molecular configuration space, the PES is evaluated only sparsely, with local approximations constructed by estimating VMC energies and forces at sampled geometries and aggregating the resulting noisy data using Gaussian process regression. Our method enables accurate and efficient exploration of complex PES landscapes, including structure relaxation, transition-state searches, and minimum-energy pathways, for both ground and excited states. This opens the door to studying bond breaking, formation, and large structural rearrangements in systems with pronounced multi-reference character.
Excited Pfaffians: Generalized Neural Wave Functions Across Structure and State
Mar 15, 2026Neural-network wave functions in Variational Monte Carlo (VMC) have achieved great success in accurately representing both ground and excited states. However, achieving sufficient numerical accuracy in state overlaps requires increasing the number of Monte Carlo samples, and consequently the computational cost, with the number of states. We present a nearly constant sample-size approach, Multi-State Importance Sampling (MSIS), that leverages samples from all states to estimate pairwise overlap. To efficiently evaluate all states for all samples, we introduce Excited Pfaffians. Inspired by Hartree-Fock, this architecture represents many states within a single neural network. Excited Pfaffians also serve as generalized wave functions, allowing a single model to represent multi-state potential energy surfaces. On the carbon dimer, we match the $O(N_s^4)$-scaling natural excited states while training $>200\times$ faster and modeling 50\% more states. Our favorable scaling enables us to be the first to use neural networks to find all distinct energy levels of the beryllium atom. Finally, we demonstrate that a single wave function can represent excited states across various molecules.
Enhanced Diffusion Sampling: Efficient Rare Event Sampling and Free Energy Calculation with Diffusion Models
Feb 18, 2026The rare-event sampling problem has long been the central limiting factor in molecular dynamics (MD), especially in biomolecular simulation. Recently, diffusion models such as BioEmu have emerged as powerful equilibrium samplers that generate independent samples from complex molecular distributions, eliminating the cost of sampling rare transition events. However, a sampling problem remains when computing observables that rely on states which are rare in equilibrium, for example folding free energies. Here, we introduce enhanced diffusion sampling, enabling efficient exploration of rare-event regions while preserving unbiased thermodynamic estimators. The key idea is to perform quantitatively accurate steering protocols to generate biased ensembles and subsequently recover equilibrium statistics via exact reweighting. We instantiate our framework in three algorithms: UmbrellaDiff (umbrella sampling with diffusion models), $Δ$G-Diff (free-energy differences via tilted ensembles), and MetaDiff (a batchwise analogue for metadynamics). Across toy systems, protein folding landscapes and folding free energies, our methods achieve fast, accurate, and scalable estimation of equilibrium properties within GPU-minutes to hours per system -- closing the rare-event sampling gap that remained after the advent of diffusion-model equilibrium samplers.
Learning Hamiltonian Flow Maps: Mean Flow Consistency for Large-Timestep Molecular Dynamics
Jan 29, 2026Simulating the long-time evolution of Hamiltonian systems is limited by the small timesteps required for stable numerical integration. To overcome this constraint, we introduce a framework to learn Hamiltonian Flow Maps by predicting the mean phase-space evolution over a chosen time span $Δt$, enabling stable large-timestep updates far beyond the stability limits of classical integrators. To this end, we impose a Mean Flow consistency condition for time-averaged Hamiltonian dynamics. Unlike prior approaches, this allows training on independent phase-space samples without access to future states, avoiding expensive trajectory generation. Validated across diverse Hamiltonian systems, our method in particular improves upon molecular dynamics simulations using machine-learned force fields (MLFF). Our models maintain comparable training and inference cost, but support significantly larger integration timesteps while trained directly on widely-available trajectory-free MLFF datasets.
Assessing generative modeling approaches for free energy estimates in condensed matter
Dec 30, 2025The accurate estimation of free energy differences between two states is a long-standing challenge in molecular simulations. Traditional approaches generally rely on sampling multiple intermediate states to ensure sufficient overlap in phase space and are, consequently, computationally expensive. Several generative-model-based methods have recently addressed this challenge by learning a direct bridge between distributions, bypassing the need for intermediate states. However, it remains unclear which approaches provide the best trade-off between efficiency, accuracy, and scalability. In this work, we systematically review these methods and benchmark selected approaches with a focus on condensed-matter systems. In particular, we investigate the performance of discrete and continuous normalizing flows in the context of targeted free energy perturbation as well as FEAT (Free energy Estimators with Adaptive Transport) together with the escorted Jarzynski equality, using coarse-grained monatomic ice and Lennard-Jones solids as benchmark systems. We evaluate accuracy, data efficiency, computational cost, and scalability with system size. Our results provide a quantitative framework for selecting effective free energy estimation strategies in condensed-phase systems.
Operator Forces For Coarse-Grained Molecular Dynamics
Jun 24, 2025Coarse-grained (CG) molecular dynamics simulations extend the length and time scale of atomistic simulations by replacing groups of correlated atoms with CG beads. Machine-learned coarse-graining (MLCG) has recently emerged as a promising approach to construct highly accurate force fields for CG molecular dynamics. However, the calibration of MLCG force fields typically hinges on force matching, which demands extensive reference atomistic trajectories with corresponding force labels. In practice, atomistic forces are often not recorded, making traditional force matching infeasible on pre-existing datasets. Recently, noise-based kernels have been introduced to adapt force matching to the low-data regime, including situations in which reference atomistic forces are not present. While this approach produces force fields which recapitulate slow collective motion, it introduces significant local distortions due to the corrupting effects of the noise-based kernel. In this work, we introduce more general kernels based on normalizing flows that substantially reduce these local distortions while preserving global conformational accuracy. We demonstrate our method on small proteins, showing that flow-based kernels can generate high-quality CG forces solely from configurational samples.
Ab-initio simulation of excited-state potential energy surfaces with transferable deep quantum Monte Carlo
Mar 25, 2025The accurate quantum chemical calculation of excited states is a challenging task, often requiring computationally demanding methods. When entire ground and excited potential energy surfaces (PESs) are desired, e.g., to predict the interaction of light excitation and structural changes, one is often forced to use cheaper computational methods at the cost of reduced accuracy. Here we introduce a novel method for the geometrically transferable optimization of neural network wave functions that leverages weight sharing and dynamical ordering of electronic states. Our method enables the efficient prediction of ground and excited-state PESs and their intersections at the highest accuracy, demonstrating up to two orders of magnitude cost reduction compared to single-point calculations. We validate our approach on three challenging excited-state PESs, including ethylene, the carbon dimer, and the methylenimmonium cation, indicating that transferable deep-learning QMC can pave the way towards highly accurate simulation of excited-state dynamics.
Doob's Lagrangian: A Sample-Efficient Variational Approach to Transition Path Sampling
Oct 10, 2024

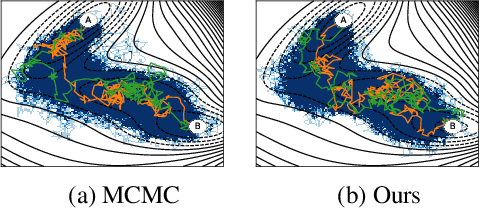
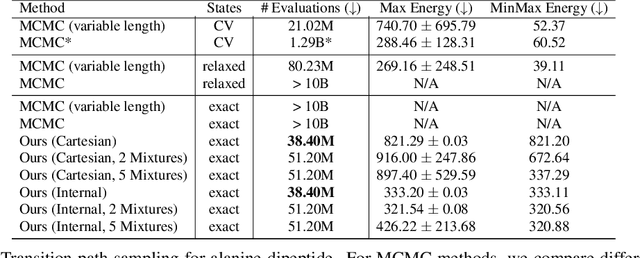
Rare event sampling in dynamical systems is a fundamental problem arising in the natural sciences, which poses significant computational challenges due to an exponentially large space of trajectories. For settings where the dynamical system of interest follows a Brownian motion with known drift, the question of conditioning the process to reach a given endpoint or desired rare event is definitively answered by Doob's h-transform. However, the naive estimation of this transform is infeasible, as it requires simulating sufficiently many forward trajectories to estimate rare event probabilities. In this work, we propose a variational formulation of Doob's $h$-transform as an optimization problem over trajectories between a given initial point and the desired ending point. To solve this optimization, we propose a simulation-free training objective with a model parameterization that imposes the desired boundary conditions by design. Our approach significantly reduces the search space over trajectories and avoids expensive trajectory simulation and inefficient importance sampling estimators which are required in existing methods. We demonstrate the ability of our method to find feasible transition paths on real-world molecular simulation and protein folding tasks.
Highly Accurate Real-space Electron Densities with Neural Networks
Sep 02, 2024Variational ab-initio methods in quantum chemistry stand out among other methods in providing direct access to the wave function. This allows in principle straightforward extraction of any other observable of interest, besides the energy, but in practice this extraction is often technically difficult and computationally impractical. Here, we consider the electron density as a central observable in quantum chemistry and introduce a novel method to obtain accurate densities from real-space many-electron wave functions by representing the density with a neural network that captures known asymptotic properties and is trained from the wave function by score matching and noise-contrastive estimation. We use variational quantum Monte Carlo with deep-learning ans\"atze (deep QMC) to obtain highly accurate wave functions free of basis set errors, and from them, using our novel method, correspondingly accurate electron densities, which we demonstrate by calculating dipole moments, nuclear forces, contact densities, and other density-based properties.