 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnabling ab initio geometry optimization of strongly correlated systems with transferable deep quantum Monte Carlo
Mar 26, 2026A faithful description of chemical processes requires exploring extended regions of the molecular potential energy surface (PES), which remains challenging for strongly correlated systems. Transferable deep-learning variational Monte Carlo (VMC) offers a promising route by efficiently solving the electronic Schrödinger equation jointly across molecular geometries at consistently high accuracy, yet its stochastic nature renders direct exploration of molecular configuration space nontrivial. Here, we present a framework for highly accurate ab initio exploration of PESs that combines transferable deep-learning VMC with a cost-effective estimation of energies, forces, and Hessians. By continuously sampling nuclear configurations during VMC optimization of electronic wave functions, we obtain transferable descriptions that achieve zero-shot chemical accuracy within chemically relevant distributions of molecular geometries. Throughout the subsequent characterization of molecular configuration space, the PES is evaluated only sparsely, with local approximations constructed by estimating VMC energies and forces at sampled geometries and aggregating the resulting noisy data using Gaussian process regression. Our method enables accurate and efficient exploration of complex PES landscapes, including structure relaxation, transition-state searches, and minimum-energy pathways, for both ground and excited states. This opens the door to studying bond breaking, formation, and large structural rearrangements in systems with pronounced multi-reference character.
Ab-initio simulation of excited-state potential energy surfaces with transferable deep quantum Monte Carlo
Mar 25, 2025The accurate quantum chemical calculation of excited states is a challenging task, often requiring computationally demanding methods. When entire ground and excited potential energy surfaces (PESs) are desired, e.g., to predict the interaction of light excitation and structural changes, one is often forced to use cheaper computational methods at the cost of reduced accuracy. Here we introduce a novel method for the geometrically transferable optimization of neural network wave functions that leverages weight sharing and dynamical ordering of electronic states. Our method enables the efficient prediction of ground and excited-state PESs and their intersections at the highest accuracy, demonstrating up to two orders of magnitude cost reduction compared to single-point calculations. We validate our approach on three challenging excited-state PESs, including ethylene, the carbon dimer, and the methylenimmonium cation, indicating that transferable deep-learning QMC can pave the way towards highly accurate simulation of excited-state dynamics.
Highly Accurate Real-space Electron Densities with Neural Networks
Sep 02, 2024Variational ab-initio methods in quantum chemistry stand out among other methods in providing direct access to the wave function. This allows in principle straightforward extraction of any other observable of interest, besides the energy, but in practice this extraction is often technically difficult and computationally impractical. Here, we consider the electron density as a central observable in quantum chemistry and introduce a novel method to obtain accurate densities from real-space many-electron wave functions by representing the density with a neural network that captures known asymptotic properties and is trained from the wave function by score matching and noise-contrastive estimation. We use variational quantum Monte Carlo with deep-learning ans\"atze (deep QMC) to obtain highly accurate wave functions free of basis set errors, and from them, using our novel method, correspondingly accurate electron densities, which we demonstrate by calculating dipole moments, nuclear forces, contact densities, and other density-based properties.
Electronic excited states in deep variational Monte Carlo
Mar 17, 2022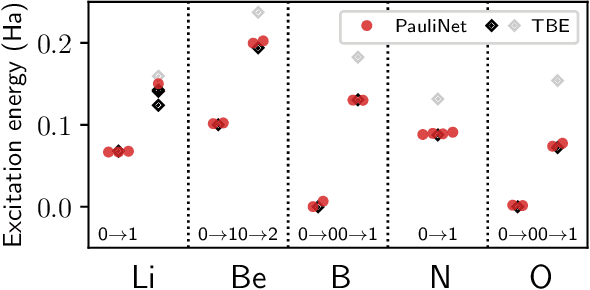
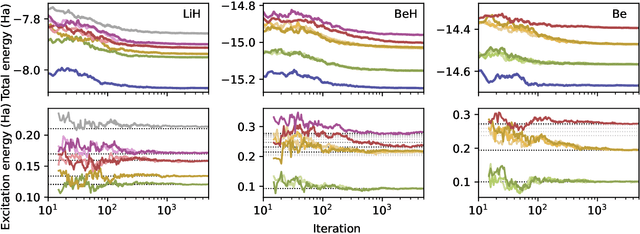
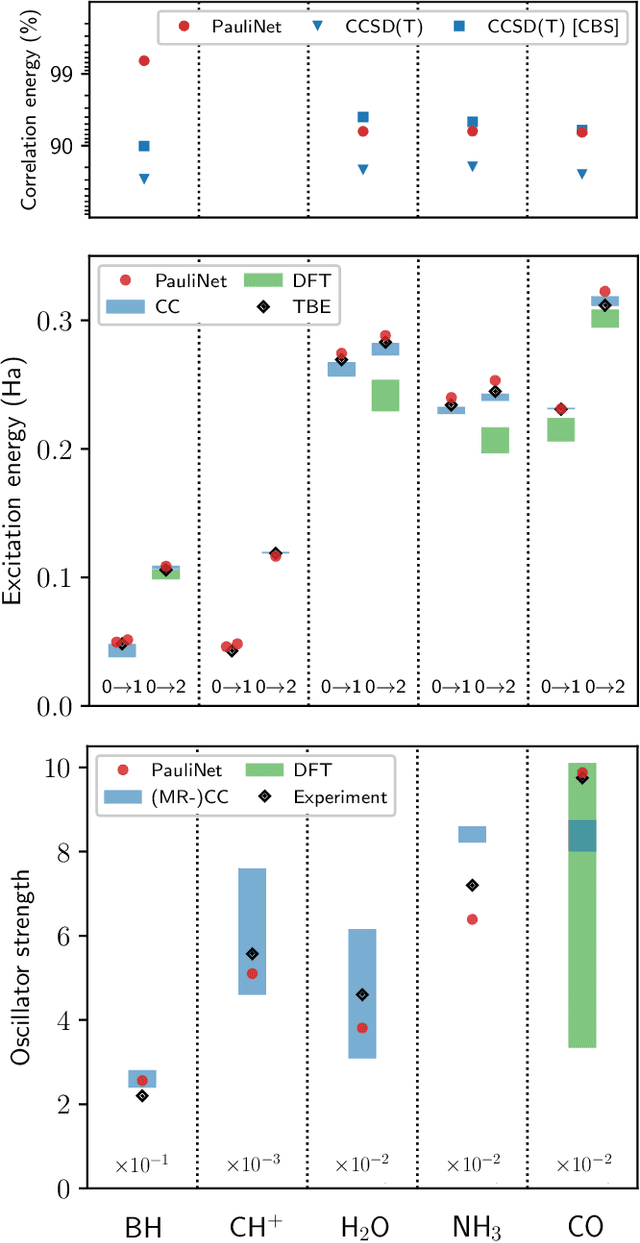
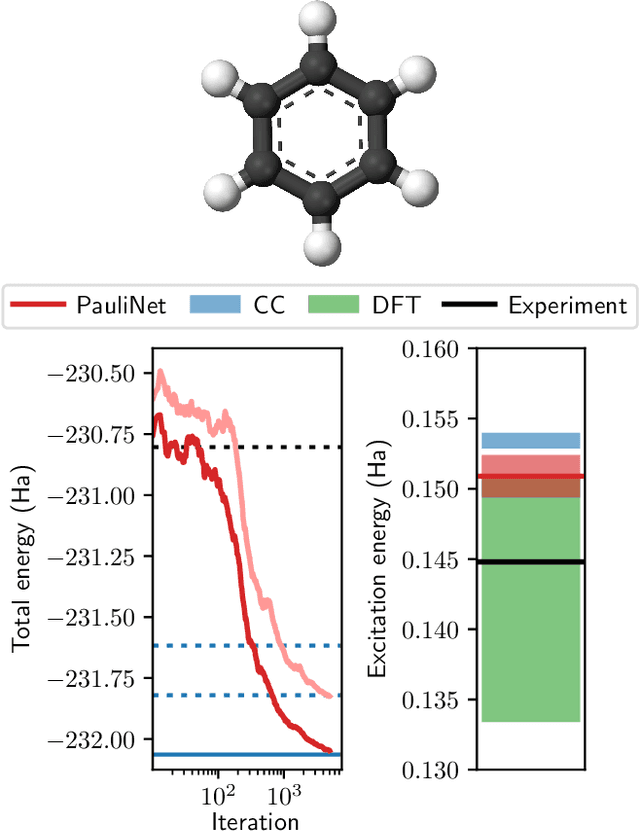
Obtaining accurate ground and low-lying excited states of electronic systems is crucial in a multitude of important applications. One ab initio method for solving the electronic Schr\"odinger equation that scales favorably for large systems and whose accuracy is limited only by the choice of wavefunction ansatz employed is variational quantum Monte Carlo (QMC). The recently introduced deep QMC approach, using a new class of ansatzes represented by deep neural networks, has been shown to generate nearly exact ground-state solutions for molecules containing up to a few dozen electrons, with the potential to scale to much larger systems where other highly accurate methods are not feasible. In this paper, we advance one such ansatz (PauliNet) to compute electronic excited states through a simple variational procedure. We demonstrate our method on a variety of small atoms and molecules where we consistently achieve high accuracy for low-lying states. To highlight the method's potential for larger systems, we show that for the benzene molecule, PauliNet is on par with significantly more expensive high-level electronic structure methods in terms of the excitation energy and outperforms them in terms of absolute energies.
Convergence to the fixed-node limit in deep variational Monte Carlo
Oct 11, 2020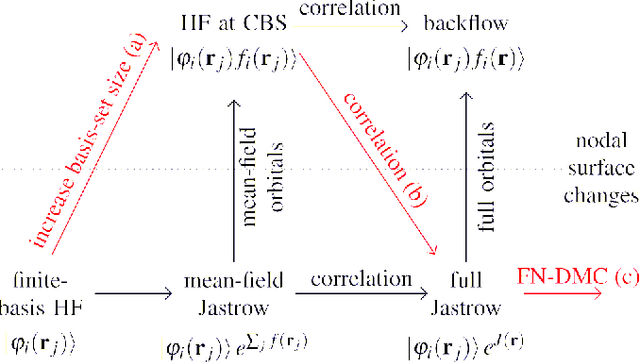
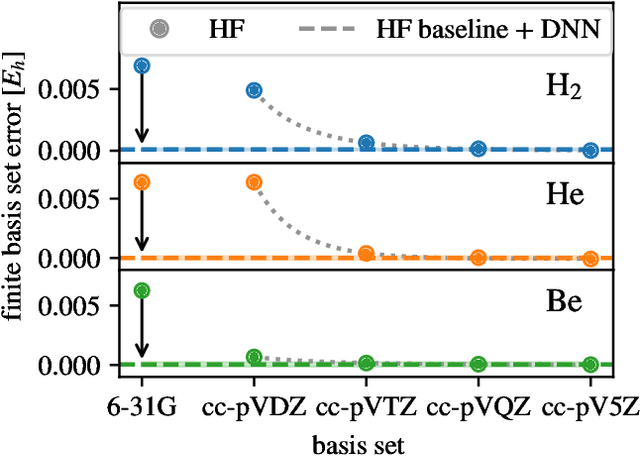
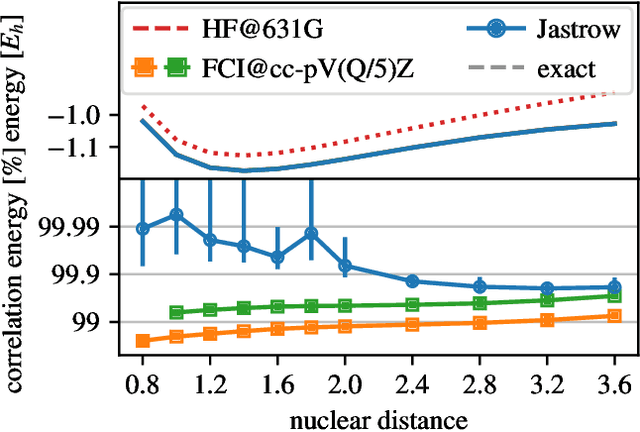
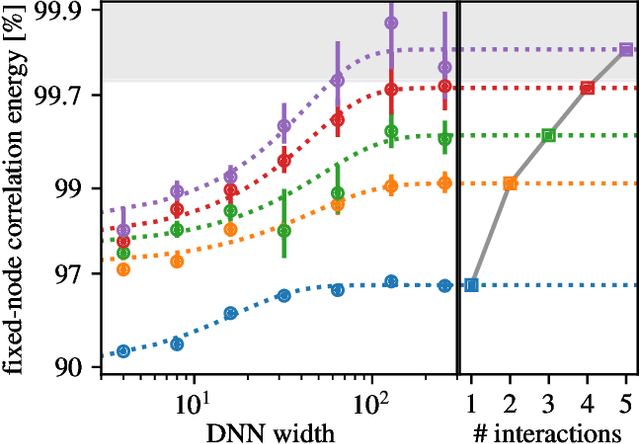
Variational quantum Monte Carlo (QMC) is an ab-initio method for solving the electronic Schr\"odinger equation that is exact in principle, but limited by the flexibility of the available ansatzes in practice. The recently introduced deep QMC approach, specifically two deep-neural-network ansatzes PauliNet and FermiNet, allows variational QMC to reach the accuracy of diffusion QMC, but little is understood about the convergence behavior of such ansatzes. Here, we analyze how deep variational QMC approaches the fixed-node limit with increasing network size. First, we demonstrate that a deep neural network can overcome the limitations of a small basis set and reach the mean-field complete-basis-set limit. Moving to electron correlation, we then perform an extensive hyperparameter scan of a deep Jastrow factor for LiH and H$_4$ and find that variational energies at the fixed-node limit can be obtained with a sufficiently large network. Finally, we benchmark mean-field and many-body ansatzes on H$_2$O, increasing the fraction of recovered fixed-node correlation energy by half an order of magnitude compared to previous VMC results. This analysis helps understanding the superb performance of deep variational ansatzes, and will guide future improvements of the neural network architectures in deep QMC.
Deep neural network solution of the electronic Schrödinger equation
Sep 16, 2019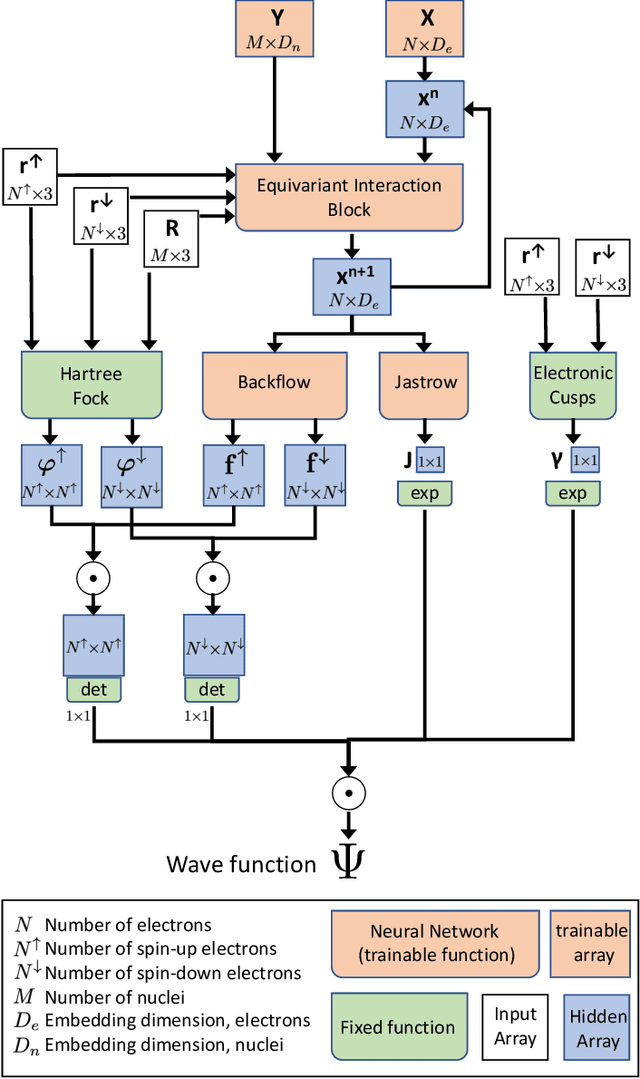

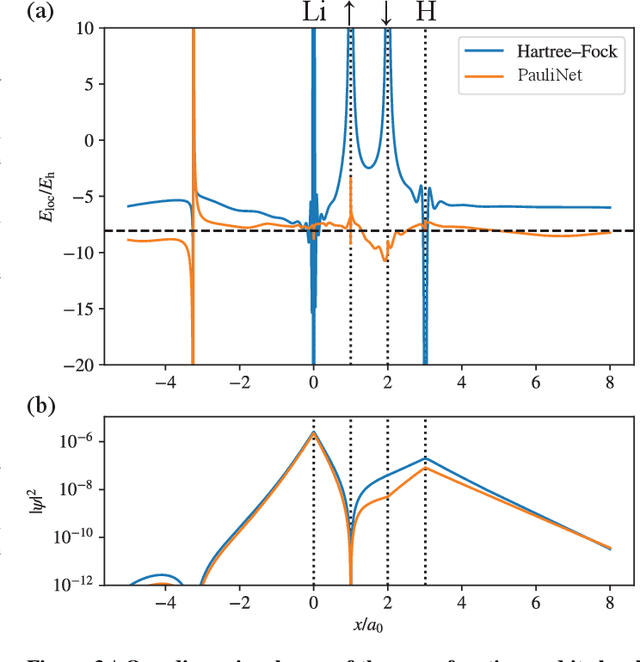
The electronic Schr\"odinger equation describes fundamental properties of molecules and materials, but cannot be solved exactly for larger systems than the hydrogen atom. Quantum Monte Carlo is a suitable method when high-quality approximations are sought, and its accuracy is in principle limited only by the flexibility of the used wave-function ansatz. Here we develop a deep-learning wave-function ansatz, dubbed PauliNet, which has the Hartree-Fock solution built in as a baseline, incorporates the physics of valid wave functions, and is trained using variational quantum Monte Carlo (VMC). Our deep-learning method achieves higher accuracy than comparable state-of-the-art VMC ansatzes for atoms, diatomic molecules and a strongly-correlated hydrogen chain. We anticipate that this method can reveal new physical insights and provide guidance for the design of molecules and materials where highly accurate quantum-mechanical solutions are needed, such as in transition metals and other strongly correlated systems.