 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScalable Machine Learning Force Fields for Macromolecular Systems Through Long-Range Aware Message Passing
Jan 07, 2026Machine learning force fields (MLFFs) have revolutionized molecular simulations by providing quantum mechanical accuracy at the speed of molecular mechanical computations. However, a fundamental reliance of these models on fixed-cutoff architectures limits their applicability to macromolecular systems where long-range interactions dominate. We demonstrate that this locality constraint causes force prediction errors to scale monotonically with system size, revealing a critical architectural bottleneck. To overcome this, we establish the systematically designed MolLR25 ({Mol}ecules with {L}ong-{R}ange effect) benchmark up to 1200 atoms, generated using high-fidelity DFT, and introduce E2Former-LSR, an equivariant transformer that explicitly integrates long-range attention blocks. E2Former-LSR exhibits stable error scaling, achieves superior fidelity in capturing non-covalent decay, and maintains precision on complex protein conformations. Crucially, its efficient design provides up to 30% speedup compared to purely local models. This work validates the necessity of non-local architectures for generalizable MLFFs, enabling high-fidelity molecular dynamics for large-scale chemical and biological systems.
Highly Accurate Real-space Electron Densities with Neural Networks
Sep 02, 2024Variational ab-initio methods in quantum chemistry stand out among other methods in providing direct access to the wave function. This allows in principle straightforward extraction of any other observable of interest, besides the energy, but in practice this extraction is often technically difficult and computationally impractical. Here, we consider the electron density as a central observable in quantum chemistry and introduce a novel method to obtain accurate densities from real-space many-electron wave functions by representing the density with a neural network that captures known asymptotic properties and is trained from the wave function by score matching and noise-contrastive estimation. We use variational quantum Monte Carlo with deep-learning ans\"atze (deep QMC) to obtain highly accurate wave functions free of basis set errors, and from them, using our novel method, correspondingly accurate electron densities, which we demonstrate by calculating dipole moments, nuclear forces, contact densities, and other density-based properties.
Error-mitigated Quantum Approximate Optimization via Learning-based Adaptive Optimization
Mar 27, 2023Combinatorial optimization problems are ubiquitous and computationally hard to solve in general. Quantum computing is envisioned as a powerful tool offering potential computational advantages for solving some of these problems. Quantum approximate optimization algorithm (QAOA), one of the most representative quantum-classical hybrid algorithms, is designed to solve certain combinatorial optimization problems by transforming a discrete optimization problem into a classical optimization problem over a continuous circuit parameter domain. QAOA objective landscape over the parameter variables is notorious for pervasive local minima and barren plateaus, and its viability in training significantly relies on the efficacy of the classical optimization algorithm. To enhance the performance of QAOA, we design double adaptive-region Bayesian optimization (DARBO), an adaptive classical optimizer for QAOA. Our experimental results demonstrate that the algorithm greatly outperforms conventional gradient-based and gradient-free optimizers in terms of speed, accuracy, and stability. We also address the issues of measurement efficiency and the suppression of quantum noise by successfully conducting the full optimization loop on the superconducting quantum processor. This work helps to unlock the full power of QAOA and paves the way toward achieving quantum advantage in practical classical tasks.
Molecular-orbital-based Machine Learning for Open-shell and Multi-reference Systems with Kernel Addition Gaussian Process Regression
Jul 17, 2022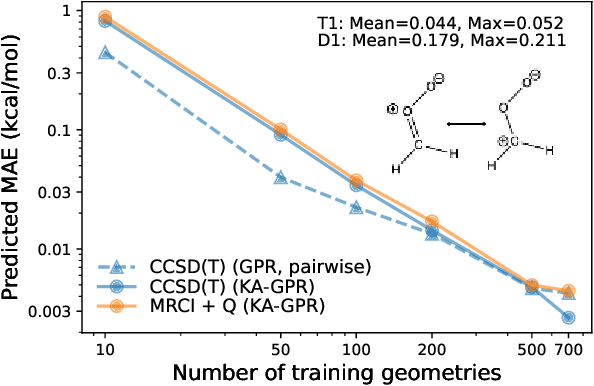
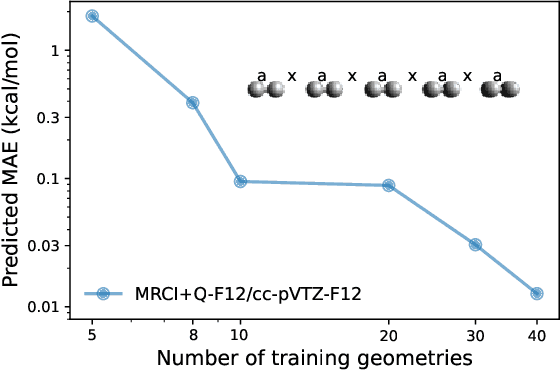
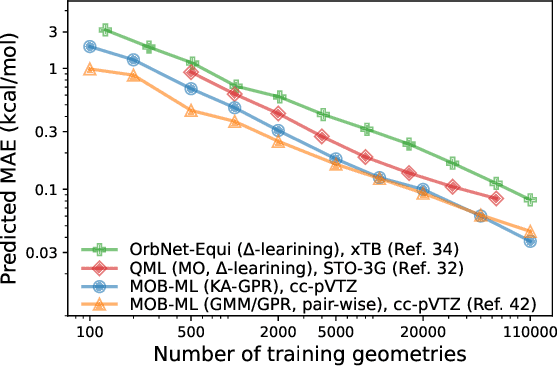
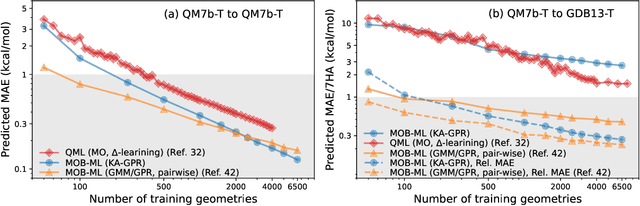
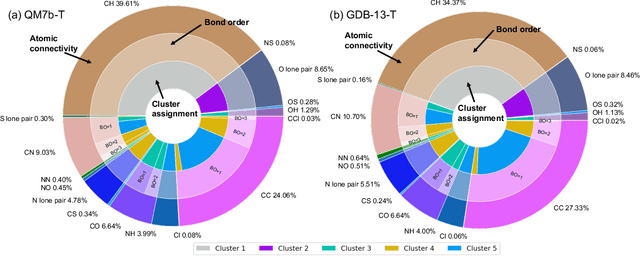
We introduce a novel machine learning strategy, kernel addition Gaussian process regression (KA-GPR), in molecular-orbital-based machine learning (MOB-ML) to learn the total correlation energies of general electronic structure theories for closed- and open-shell systems by introducing a machine learning strategy. The learning efficiency of MOB-ML (KA-GPR) is the same as the original MOB-ML method for the smallest criegee molecule, which is a closed-shell molecule with multi-reference characters. In addition, the prediction accuracies of different small free radicals could reach the chemical accuracy of 1 kcal/mol by training on one example structure. Accurate potential energy surfaces for the H10 chain (closed-shell) and water OH bond dissociation (open-shell) could also be generated by MOB-ML (KA-GPR). To explore the breadth of chemical systems that KA-GPR can describe, we further apply MOB-ML to accurately predict the large benchmark datasets for closed- (QM9, QM7b-T, GDB-13-T) and open-shell (QMSpin) molecules.
Molecular Dipole Moment Learning via Rotationally Equivariant Gaussian Process Regression with Derivatives in Molecular-orbital-based Machine Learning
May 31, 2022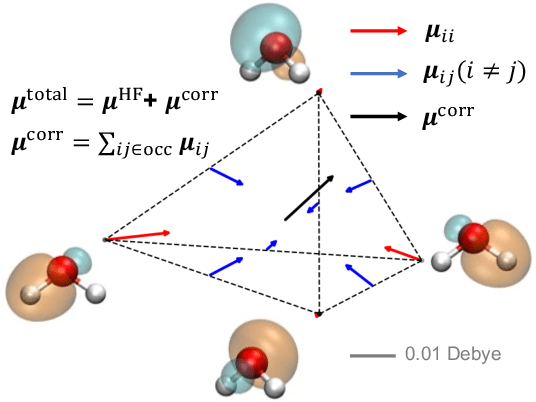

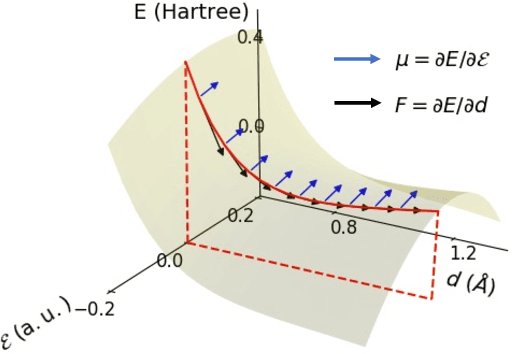
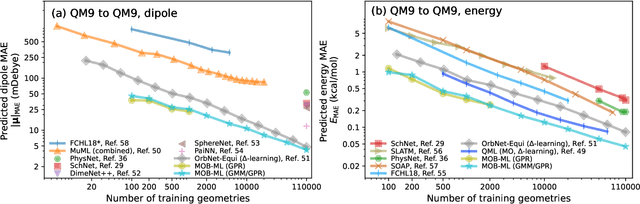
This study extends the accurate and transferable molecular-orbital-based machine learning (MOB-ML) approach to modeling the contribution of electron correlation to dipole moments at the cost of Hartree-Fock computations. A molecular-orbital-based (MOB) pairwise decomposition of the correlation part of the dipole moment is applied, and these pair dipole moments could be further regressed as a universal function of molecular orbitals (MOs). The dipole MOB features consist of the energy MOB features and their responses to electric fields. An interpretable and rotationally equivariant Gaussian process regression (GPR) with derivatives algorithm is introduced to learn the dipole moment more efficiently. The proposed problem setup, feature design, and ML algorithm are shown to provide highly-accurate models for both dipole moment and energies on water and fourteen small molecules. To demonstrate the ability of MOB-ML to function as generalized density-matrix functionals for molecular dipole moments and energies of organic molecules, we further apply the proposed MOB-ML approach to train and test the molecules from the QM9 dataset. The application of local scalable GPR with Gaussian mixture model unsupervised clustering (GMM/GPR) scales up MOB-ML to a large-data regime while retaining the prediction accuracy. In addition, compared with literature results, MOB-ML provides the best test MAEs of 4.21 mDebye and 0.045 kcal/mol for dipole moment and energy models, respectively, when training on 110000 QM9 molecules. The excellent transferability of the resulting QM9 models is also illustrated by the accurate predictions for four different series of peptides.
ODBO: Bayesian Optimization with Search Space Prescreening for Directed Protein Evolution
May 20, 2022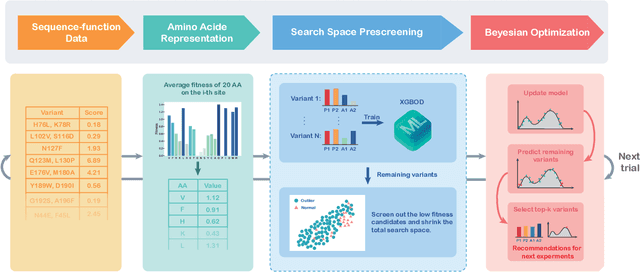

Directed evolution is a versatile technique in protein engineering that mimics the process of natural selection by iteratively alternating between mutagenesis and screening in order to search for sequences that optimize a given property of interest, such as catalytic activity and binding affinity to a specified target. However, the space of possible proteins is too large to search exhaustively in the laboratory, and functional proteins are scarce in the vast sequence space. Machine learning (ML) approaches can accelerate directed evolution by learning to map protein sequences to functions without building a detailed model of the underlying physics, chemistry and biological pathways. Despite the great potentials held by these ML methods, they encounter severe challenges in identifying the most suitable sequences for a targeted function. These failures can be attributed to the common practice of adopting a high-dimensional feature representation for protein sequences and inefficient search methods. To address these issues, we propose an efficient, experimental design-oriented closed-loop optimization framework for protein directed evolution, termed ODBO, which employs a combination of novel low-dimensional protein encoding strategy and Bayesian optimization enhanced with search space prescreening via outlier detection. We further design an initial sample selection strategy to minimize the number of experimental samples for training ML models. We conduct and report four protein directed evolution experiments that substantiate the capability of the proposed framework for finding of the variants with properties of interest. We expect the ODBO framework to greatly reduce the experimental cost and time cost of directed evolution, and can be further generalized as a powerful tool for adaptive experimental design in a broader context.
Accurate Molecular-Orbital-Based Machine Learning Energies via Unsupervised Clustering of Chemical Space
Apr 21, 2022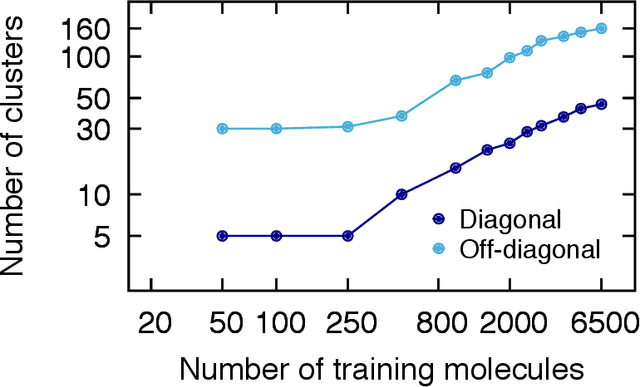
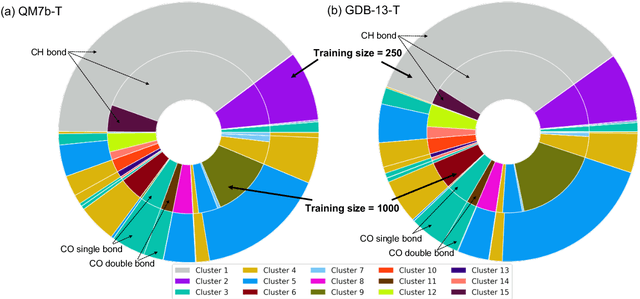
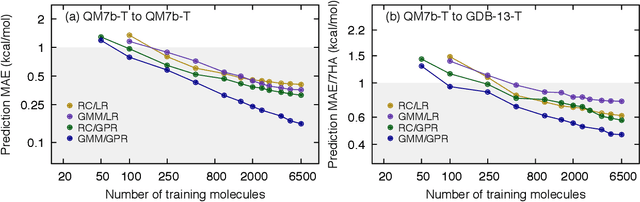
We introduce an unsupervised clustering algorithm to improve training efficiency and accuracy in predicting energies using molecular-orbital-based machine learning (MOB-ML). This work determines clusters via the Gaussian mixture model (GMM) in an entirely automatic manner and simplifies an earlier supervised clustering approach [J. Chem. Theory Comput., 15, 6668 (2019)] by eliminating both the necessity for user-specified parameters and the training of an additional classifier. Unsupervised clustering results from GMM have the advantage of accurately reproducing chemically intuitive groupings of frontier molecular orbitals and having improved performance with an increasing number of training examples. The resulting clusters from supervised or unsupervised clustering is further combined with scalable Gaussian process regression (GPR) or linear regression (LR) to learn molecular energies accurately by generating a local regression model in each cluster. Among all four combinations of regressors and clustering methods, GMM combined with scalable exact Gaussian process regression (GMM/GPR) is the most efficient training protocol for MOB-ML. The numerical tests of molecular energy learning on thermalized datasets of drug-like molecules demonstrate the improved accuracy, transferability, and learning efficiency of GMM/GPR over not only other training protocols for MOB-ML, i.e., supervised regression-clustering combined with GPR(RC/GPR) and GPR without clustering. GMM/GPR also provide the best molecular energy predictions compared with the ones from literature on the same benchmark datasets. With a lower scaling, GMM/GPR has a 10.4-fold speedup in wall-clock training time compared with scalable exact GPR with a training size of 6500 QM7b-T molecules.
Molecular Energy Learning Using Alternative Blackbox Matrix-Matrix Multiplication Algorithm for Exact Gaussian Process
Sep 20, 2021
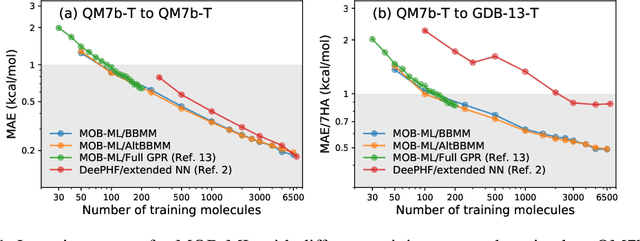

We present an application of the blackbox matrix-matrix multiplication (BBMM) algorithm to scale up the Gaussian Process (GP) training of molecular energies in the molecular-orbital based machine learning (MOB-ML) framework. An alternative implementation of BBMM (AltBBMM) is also proposed to train more efficiently (over four-fold speedup) with the same accuracy and transferability as the original BBMM implementation. The training of MOB-ML was limited to 220 molecules, and BBMM and AltBBMM scale the training of MOB-ML up by over 30 times to 6500 molecules (more than a million pair energies). The accuracy and transferability of both algorithms are examined on the benchmark datasets of organic molecules with 7 and 13 heavy atoms. These lower-scaling implementations of the GP preserve the state-of-the-art learning efficiency in the low-data regime while extending it to the large-data regime with better accuracy than other available machine learning works on molecular energies.
Regression-clustering for Improved Accuracy and Training Cost with Molecular-Orbital-Based Machine Learning
Sep 09, 2019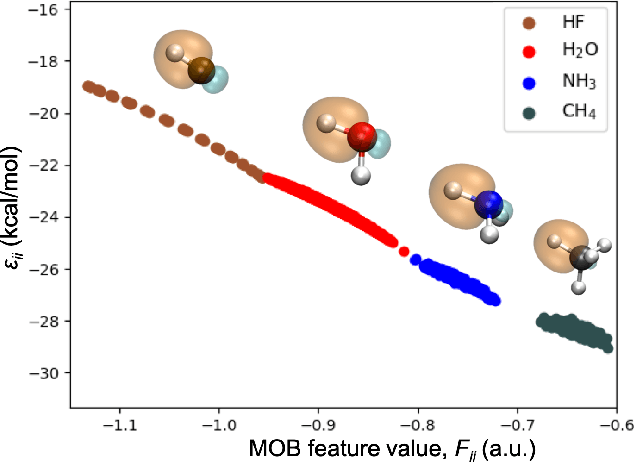
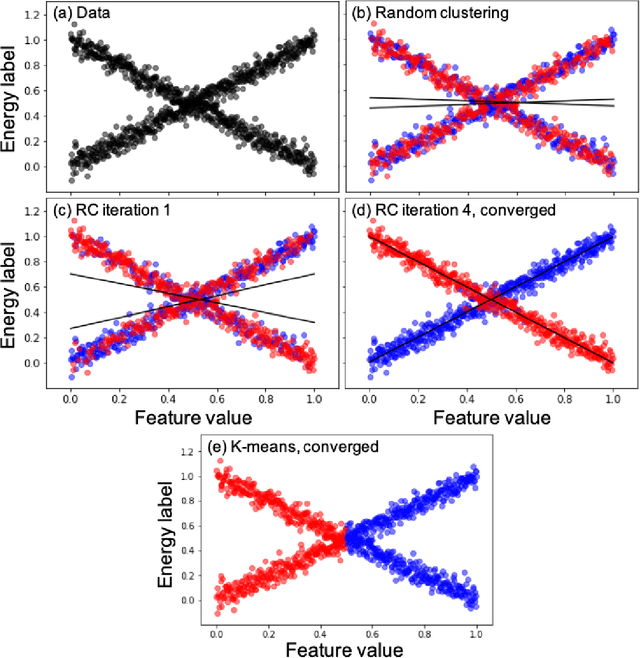
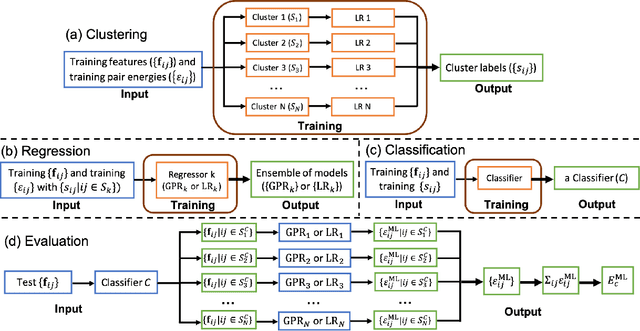
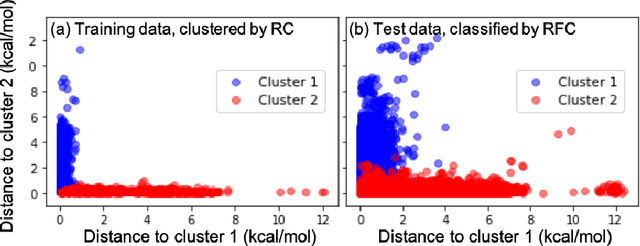
Machine learning (ML) in the representation of molecular-orbital-based (MOB) features has been shown to be an accurate and transferable approach to the prediction of post-Hartree-Fock correlation energies. Previous applications of MOB-ML employed Gaussian Process Regression (GPR), which provides good prediction accuracy with small training sets; however, the cost of GPR training scales cubically with the amount of data and becomes a computational bottleneck for large training sets. In the current work, we address this problem by introducing a clustering/regression/classification implementation of MOB-ML. In a first step, regression clustering (RC) is used to partition the training data to best fit an ensemble of linear regression (LR) models; in a second step, each cluster is regressed independently, using either LR or GPR; and in a third step, a random forest classifier (RFC) is trained for the prediction of cluster assignments based on MOB feature values. Upon inspection, RC is found to recapitulate chemically intuitive groupings of the frontier molecular orbitals, and the combined RC/LR/RFC and RC/GPR/RFC implementations of MOB-ML are found to provide good prediction accuracy with greatly reduced wall-clock training times. For a dataset of thermalized geometries of 7211 organic molecules of up to seven heavy atoms, both implementations reach chemical accuracy (1 kcal/mol error) with only 300 training molecules, while providing 35000-fold and 4500-fold reductions in the wall-clock training time, respectively, compared to MOB-ML without clustering. The resulting models are also demonstrated to retain transferability for the prediction of large-molecule energies with only small-molecule training data. Finally, it is shown that capping the number of training datapoints per cluster leads to further improvements in prediction accuracy with negligible increases in wall-clock training time.
A Universal Density Matrix Functional from Molecular Orbital-Based Machine Learning: Transferability across Organic Molecules
Jan 10, 2019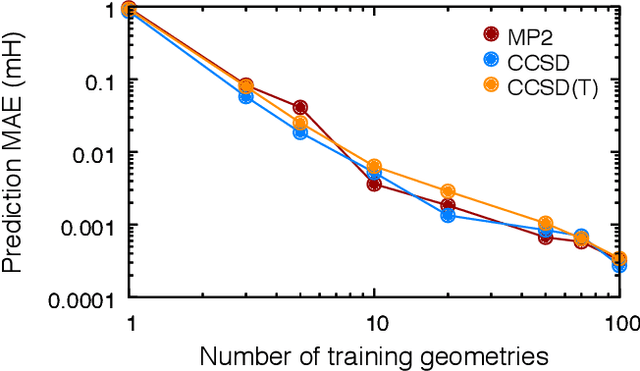
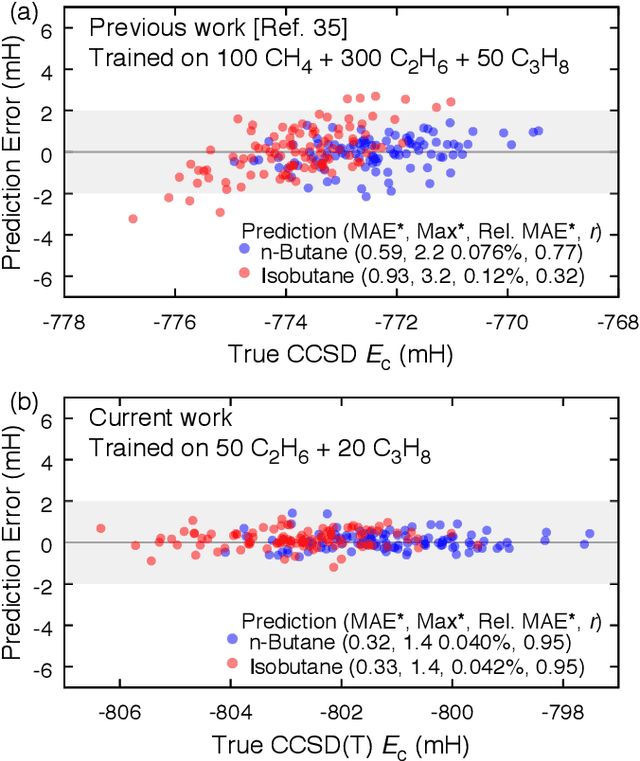
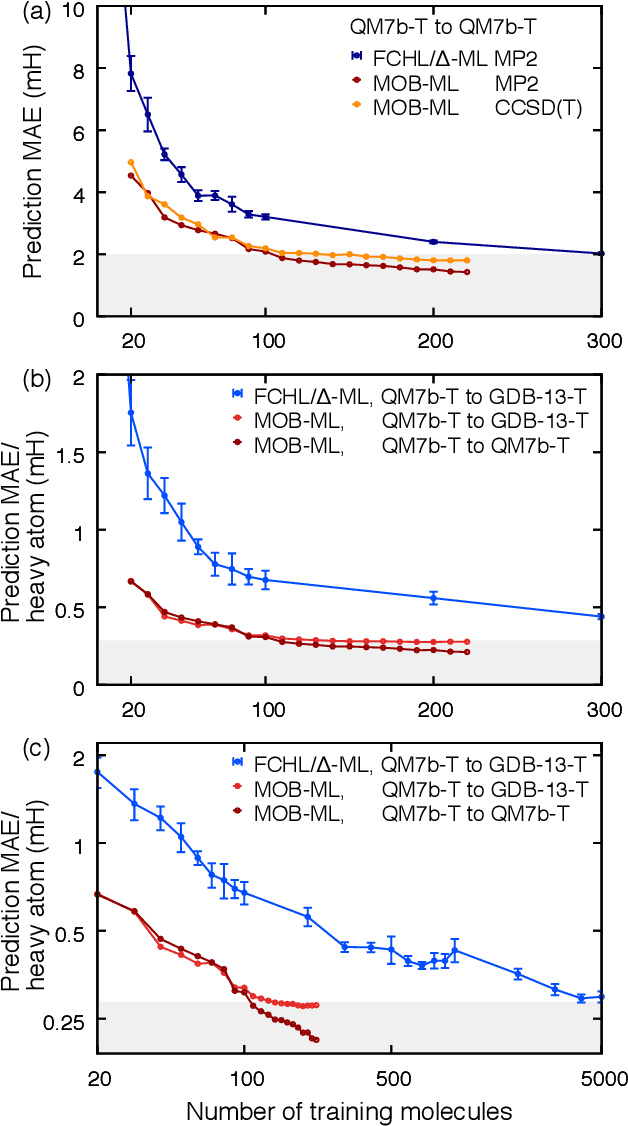
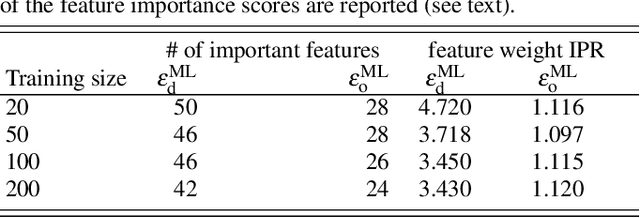
We address the degree to which machine learning can be used to accurately and transferably predict post-Hartree-Fock correlation energies. After presenting refined strategies for feature design and selection, the molecular-orbital-based machine learning (MOB-ML) method is first applied to benchmark test systems. It is shown that the total electronic energy for a set of 1000 randomized geometries of water can be described to within 1 millihartree using a model that is trained at the level of MP2, CCSD, or CCSD(T) using only a single reference calculation at a randomized geometry. To explore the breadth of chemical diversity that can be described, the MOB-ML method is then applied a set of 7211 organic models with up to seven heavy atoms. It is shown that MP2 calculations on only 90 molecules are needed to train a model that predicts MP2 energies to within 2 millihartree accuracy for the remaining 7121 molecules; likewise, CCSD(T) calculations on only 150 molecules are needed to train a model that predicts CCSD(T) energies for the remaining molecules to within 2 millihartree accuracy. The MP2 model, trained with only 90 reference calculations on seven-heavy-atom molecules, is then applied to a diverse set of 1000 thirteen-heavy-atom organic molecules, demonstrating transferable preservation of chemical accuracy.