 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecular-orbital-based Machine Learning for Open-shell and Multi-reference Systems with Kernel Addition Gaussian Process Regression
Jul 17, 2022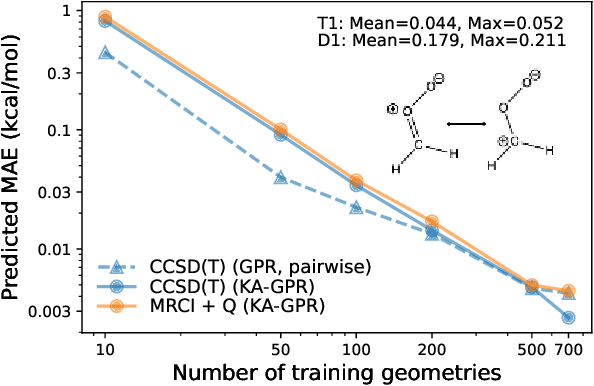
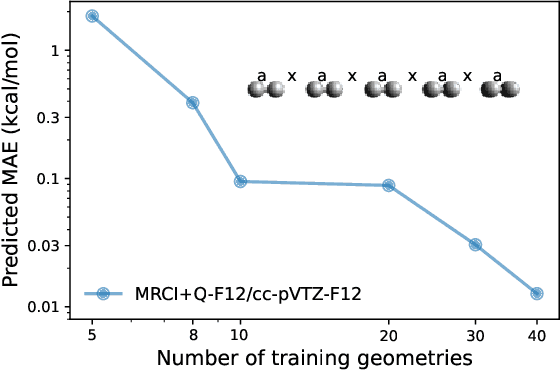
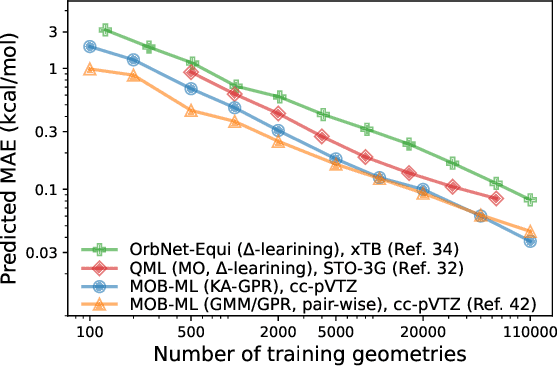
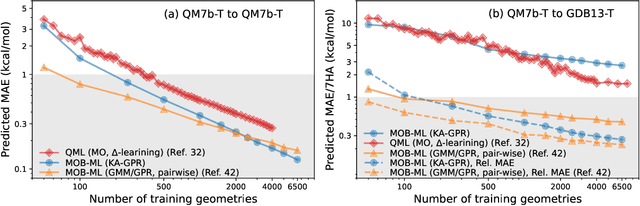
We introduce a novel machine learning strategy, kernel addition Gaussian process regression (KA-GPR), in molecular-orbital-based machine learning (MOB-ML) to learn the total correlation energies of general electronic structure theories for closed- and open-shell systems by introducing a machine learning strategy. The learning efficiency of MOB-ML (KA-GPR) is the same as the original MOB-ML method for the smallest criegee molecule, which is a closed-shell molecule with multi-reference characters. In addition, the prediction accuracies of different small free radicals could reach the chemical accuracy of 1 kcal/mol by training on one example structure. Accurate potential energy surfaces for the H10 chain (closed-shell) and water OH bond dissociation (open-shell) could also be generated by MOB-ML (KA-GPR). To explore the breadth of chemical systems that KA-GPR can describe, we further apply MOB-ML to accurately predict the large benchmark datasets for closed- (QM9, QM7b-T, GDB-13-T) and open-shell (QMSpin) molecules.