 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAccurate Molecular-Orbital-Based Machine Learning Energies via Unsupervised Clustering of Chemical Space
Paper and Code
Apr 21, 2022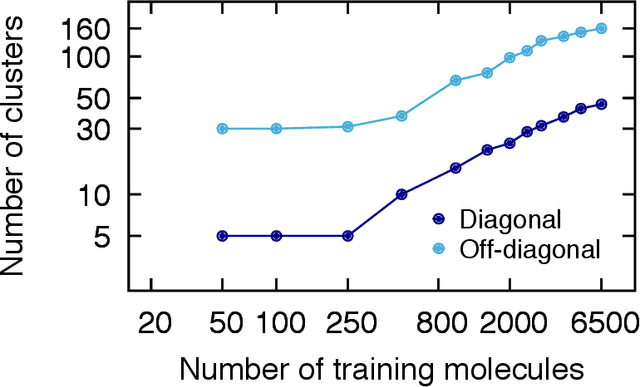
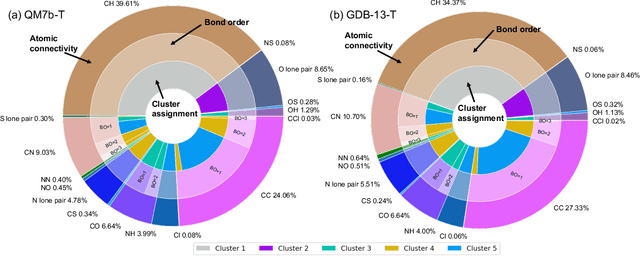
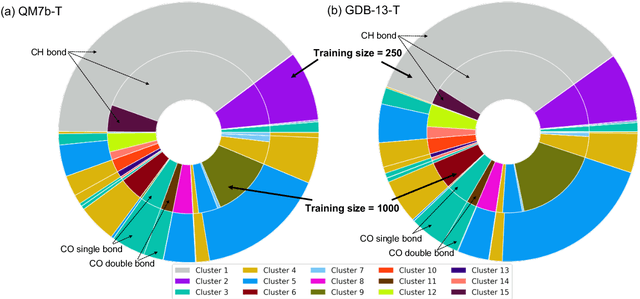
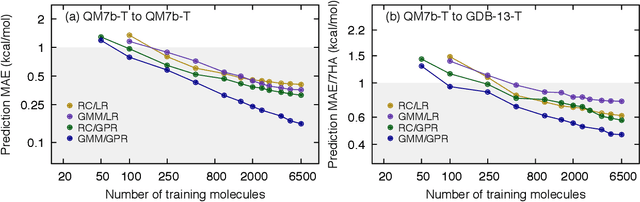
We introduce an unsupervised clustering algorithm to improve training efficiency and accuracy in predicting energies using molecular-orbital-based machine learning (MOB-ML). This work determines clusters via the Gaussian mixture model (GMM) in an entirely automatic manner and simplifies an earlier supervised clustering approach [J. Chem. Theory Comput., 15, 6668 (2019)] by eliminating both the necessity for user-specified parameters and the training of an additional classifier. Unsupervised clustering results from GMM have the advantage of accurately reproducing chemically intuitive groupings of frontier molecular orbitals and having improved performance with an increasing number of training examples. The resulting clusters from supervised or unsupervised clustering is further combined with scalable Gaussian process regression (GPR) or linear regression (LR) to learn molecular energies accurately by generating a local regression model in each cluster. Among all four combinations of regressors and clustering methods, GMM combined with scalable exact Gaussian process regression (GMM/GPR) is the most efficient training protocol for MOB-ML. The numerical tests of molecular energy learning on thermalized datasets of drug-like molecules demonstrate the improved accuracy, transferability, and learning efficiency of GMM/GPR over not only other training protocols for MOB-ML, i.e., supervised regression-clustering combined with GPR(RC/GPR) and GPR without clustering. GMM/GPR also provide the best molecular energy predictions compared with the ones from literature on the same benchmark datasets. With a lower scaling, GMM/GPR has a 10.4-fold speedup in wall-clock training time compared with scalable exact GPR with a training size of 6500 QM7b-T molecules.