 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRegression-clustering for Improved Accuracy and Training Cost with Molecular-Orbital-Based Machine Learning
Paper and Code
Sep 09, 2019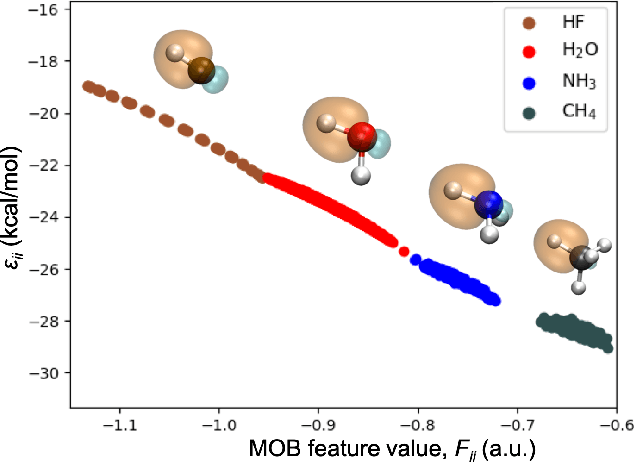
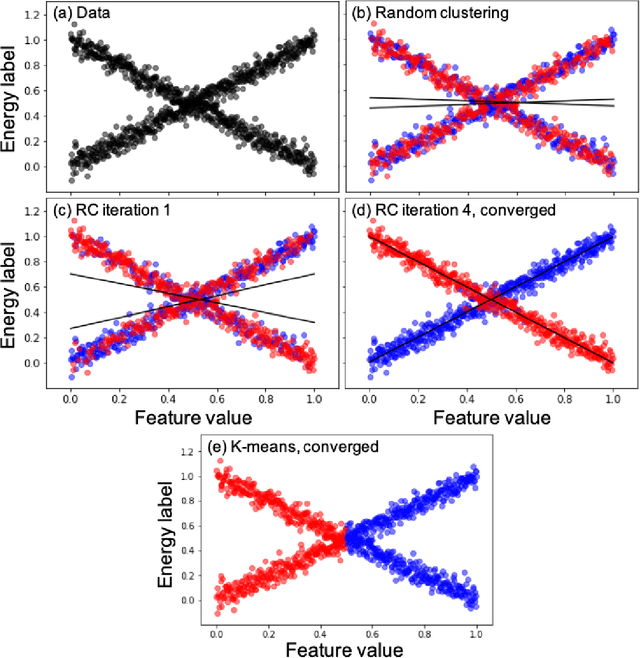
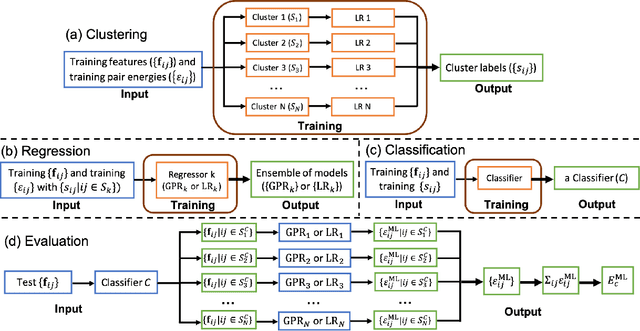
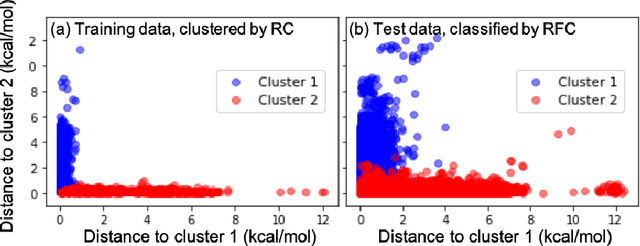
Machine learning (ML) in the representation of molecular-orbital-based (MOB) features has been shown to be an accurate and transferable approach to the prediction of post-Hartree-Fock correlation energies. Previous applications of MOB-ML employed Gaussian Process Regression (GPR), which provides good prediction accuracy with small training sets; however, the cost of GPR training scales cubically with the amount of data and becomes a computational bottleneck for large training sets. In the current work, we address this problem by introducing a clustering/regression/classification implementation of MOB-ML. In a first step, regression clustering (RC) is used to partition the training data to best fit an ensemble of linear regression (LR) models; in a second step, each cluster is regressed independently, using either LR or GPR; and in a third step, a random forest classifier (RFC) is trained for the prediction of cluster assignments based on MOB feature values. Upon inspection, RC is found to recapitulate chemically intuitive groupings of the frontier molecular orbitals, and the combined RC/LR/RFC and RC/GPR/RFC implementations of MOB-ML are found to provide good prediction accuracy with greatly reduced wall-clock training times. For a dataset of thermalized geometries of 7211 organic molecules of up to seven heavy atoms, both implementations reach chemical accuracy (1 kcal/mol error) with only 300 training molecules, while providing 35000-fold and 4500-fold reductions in the wall-clock training time, respectively, compared to MOB-ML without clustering. The resulting models are also demonstrated to retain transferability for the prediction of large-molecule energies with only small-molecule training data. Finally, it is shown that capping the number of training datapoints per cluster leads to further improvements in prediction accuracy with negligible increases in wall-clock training time.