 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeODBO: Bayesian Optimization with Search Space Prescreening for Directed Protein Evolution
May 20, 2022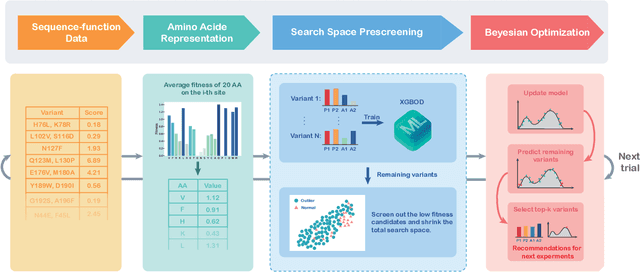
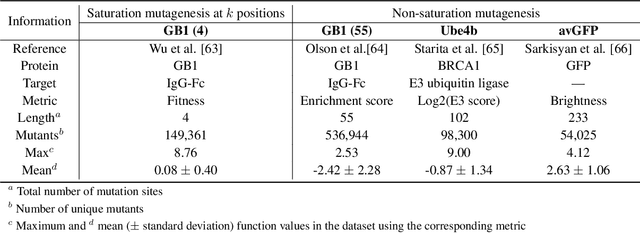
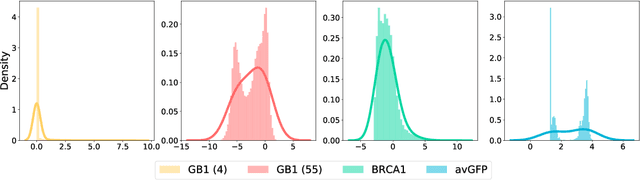

Directed evolution is a versatile technique in protein engineering that mimics the process of natural selection by iteratively alternating between mutagenesis and screening in order to search for sequences that optimize a given property of interest, such as catalytic activity and binding affinity to a specified target. However, the space of possible proteins is too large to search exhaustively in the laboratory, and functional proteins are scarce in the vast sequence space. Machine learning (ML) approaches can accelerate directed evolution by learning to map protein sequences to functions without building a detailed model of the underlying physics, chemistry and biological pathways. Despite the great potentials held by these ML methods, they encounter severe challenges in identifying the most suitable sequences for a targeted function. These failures can be attributed to the common practice of adopting a high-dimensional feature representation for protein sequences and inefficient search methods. To address these issues, we propose an efficient, experimental design-oriented closed-loop optimization framework for protein directed evolution, termed ODBO, which employs a combination of novel low-dimensional protein encoding strategy and Bayesian optimization enhanced with search space prescreening via outlier detection. We further design an initial sample selection strategy to minimize the number of experimental samples for training ML models. We conduct and report four protein directed evolution experiments that substantiate the capability of the proposed framework for finding of the variants with properties of interest. We expect the ODBO framework to greatly reduce the experimental cost and time cost of directed evolution, and can be further generalized as a powerful tool for adaptive experimental design in a broader context.
SPLDExtraTrees: Robust machine learning approach for predicting kinase inhibitor resistance
Nov 25, 2021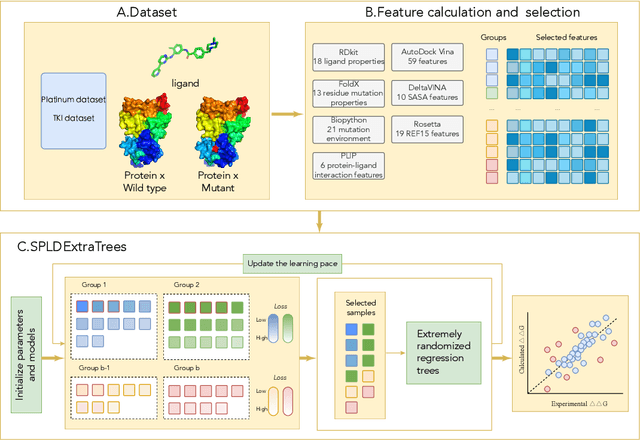
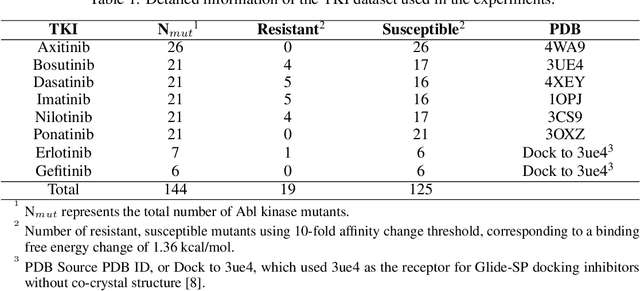
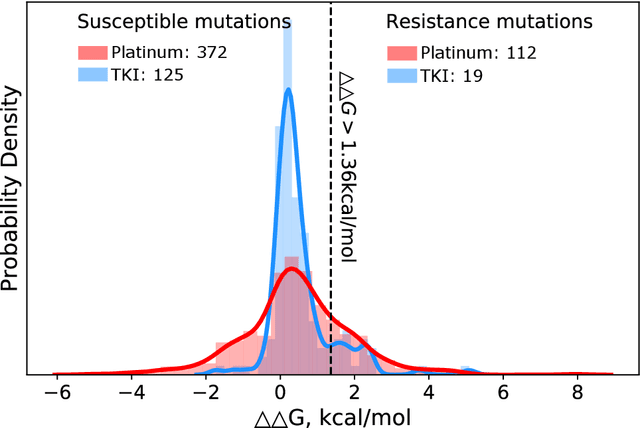
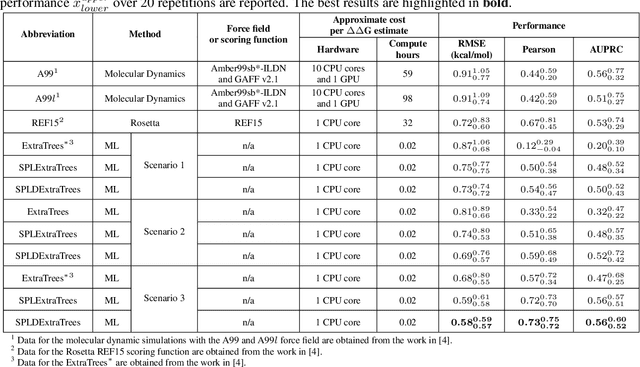
Drug resistance is a major threat to the global health and a significant concern throughout the clinical treatment of diseases and drug development. The mutation in proteins that is related to drug binding is a common cause for adaptive drug resistance. Therefore, quantitative estimations of how mutations would affect the interaction between a drug and the target protein would be of vital significance for the drug development and the clinical practice. Computational methods that rely on molecular dynamics simulations, Rosetta protocols, as well as machine learning methods have been proven to be capable of predicting ligand affinity changes upon protein mutation. However, the severely limited sample size and heavy noise induced overfitting and generalization issues have impeded wide adoption of machine learning for studying drug resistance. In this paper, we propose a robust machine learning method, termed SPLDExtraTrees, which can accurately predict ligand binding affinity changes upon protein mutation and identify resistance-causing mutations. Especially, the proposed method ranks training data following a specific scheme that starts with easy-to-learn samples and gradually incorporates harder and diverse samples into the training, and then iterates between sample weight recalculations and model updates. In addition, we calculate additional physics-based structural features to provide the machine learning model with the valuable domain knowledge on proteins for this data-limited predictive tasks. The experiments substantiate the capability of the proposed method for predicting kinase inhibitor resistance under three scenarios, and achieves predictive accuracy comparable to that of molecular dynamics and Rosetta methods with much less computational costs.