 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMulti-task Bioassay Pre-training for Protein-ligand Binding Affinity Prediction
Jun 08, 2023



Protein-ligand binding affinity (PLBA) prediction is the fundamental task in drug discovery. Recently, various deep learning-based models predict binding affinity by incorporating the three-dimensional structure of protein-ligand complexes as input and achieving astounding progress. However, due to the scarcity of high-quality training data, the generalization ability of current models is still limited. In addition, different bioassays use varying affinity measurement labels (i.e., IC50, Ki, Kd), and different experimental conditions inevitably introduce systematic noise, which poses a significant challenge to constructing high-precision affinity prediction models. To address these issues, we (1) propose Multi-task Bioassay Pre-training (MBP), a pre-training framework for structure-based PLBA prediction; (2) construct a pre-training dataset called ChEMBL-Dock with more than 300k experimentally measured affinity labels and about 2.8M docked three-dimensional structures. By introducing multi-task pre-training to treat the prediction of different affinity labels as different tasks and classifying relative rankings between samples from the same bioassay, MBP learns robust and transferrable structural knowledge from our new ChEMBL-Dock dataset with varied and noisy labels. Experiments substantiate the capability of MBP as a general framework that can improve and be tailored to mainstream structure-based PLBA prediction tasks. To the best of our knowledge, MBP is the first affinity pre-training model and shows great potential for future development.
Protein-Ligand Complex Generator & Drug Screening via Tiered Tensor Transform
Jan 03, 2023



Accurate determination of a small molecule candidate (ligand) binding pose in its target protein pocket is important for computer-aided drug discovery. Typical rigid-body docking methods ignore the pocket flexibility of protein, while the more accurate pose generation using molecular dynamics is hindered by slow protein dynamics. We develop a tiered tensor transform (3T) algorithm to rapidly generate diverse protein-ligand complex conformations for both pose and affinity estimation in drug screening, requiring neither machine learning training nor lengthy dynamics computation, while maintaining both coarse-grain-like coordinated protein dynamics and atomistic-level details of the complex pocket. The 3T conformation structures we generate are closer to experimental co-crystal structures than those generated by docking software, and more importantly achieve significantly higher accuracy in active ligand classification than traditional ensemble docking using hundreds of experimental protein conformations. 3T structure transformation is decoupled from the system physics, making future usage in other computational scientific domains possible.
SPLDExtraTrees: Robust machine learning approach for predicting kinase inhibitor resistance
Nov 25, 2021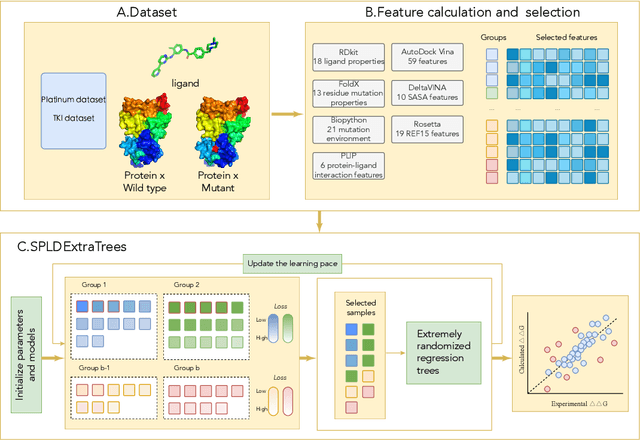
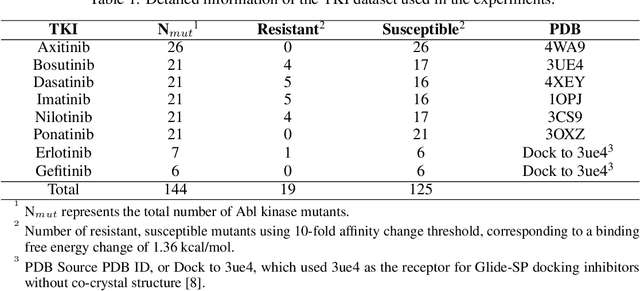
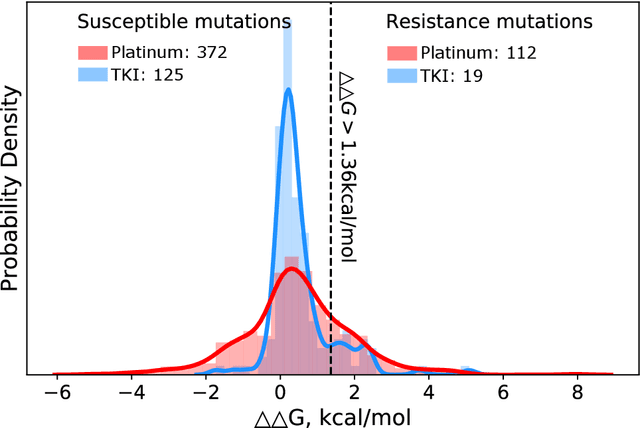
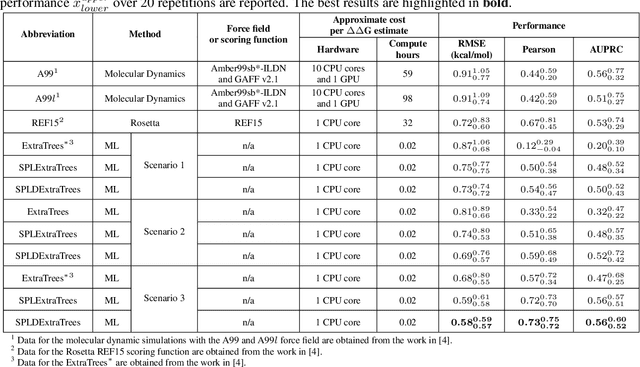
Drug resistance is a major threat to the global health and a significant concern throughout the clinical treatment of diseases and drug development. The mutation in proteins that is related to drug binding is a common cause for adaptive drug resistance. Therefore, quantitative estimations of how mutations would affect the interaction between a drug and the target protein would be of vital significance for the drug development and the clinical practice. Computational methods that rely on molecular dynamics simulations, Rosetta protocols, as well as machine learning methods have been proven to be capable of predicting ligand affinity changes upon protein mutation. However, the severely limited sample size and heavy noise induced overfitting and generalization issues have impeded wide adoption of machine learning for studying drug resistance. In this paper, we propose a robust machine learning method, termed SPLDExtraTrees, which can accurately predict ligand binding affinity changes upon protein mutation and identify resistance-causing mutations. Especially, the proposed method ranks training data following a specific scheme that starts with easy-to-learn samples and gradually incorporates harder and diverse samples into the training, and then iterates between sample weight recalculations and model updates. In addition, we calculate additional physics-based structural features to provide the machine learning model with the valuable domain knowledge on proteins for this data-limited predictive tasks. The experiments substantiate the capability of the proposed method for predicting kinase inhibitor resistance under three scenarios, and achieves predictive accuracy comparable to that of molecular dynamics and Rosetta methods with much less computational costs.