 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeKimi K2.5: Visual Agentic Intelligence
Feb 02, 2026We introduce Kimi K2.5, an open-source multimodal agentic model designed to advance general agentic intelligence. K2.5 emphasizes the joint optimization of text and vision so that two modalities enhance each other. This includes a series of techniques such as joint text-vision pre-training, zero-vision SFT, and joint text-vision reinforcement learning. Building on this multimodal foundation, K2.5 introduces Agent Swarm, a self-directed parallel agent orchestration framework that dynamically decomposes complex tasks into heterogeneous sub-problems and executes them concurrently. Extensive evaluations show that Kimi K2.5 achieves state-of-the-art results across various domains including coding, vision, reasoning, and agentic tasks. Agent Swarm also reduces latency by up to $4.5\times$ over single-agent baselines. We release the post-trained Kimi K2.5 model checkpoint to facilitate future research and real-world applications of agentic intelligence.
Kimi Linear: An Expressive, Efficient Attention Architecture
Oct 30, 2025We introduce Kimi Linear, a hybrid linear attention architecture that, for the first time, outperforms full attention under fair comparisons across various scenarios -- including short-context, long-context, and reinforcement learning (RL) scaling regimes. At its core lies Kimi Delta Attention (KDA), an expressive linear attention module that extends Gated DeltaNet with a finer-grained gating mechanism, enabling more effective use of limited finite-state RNN memory. Our bespoke chunkwise algorithm achieves high hardware efficiency through a specialized variant of the Diagonal-Plus-Low-Rank (DPLR) transition matrices, which substantially reduces computation compared to the general DPLR formulation while remaining more consistent with the classical delta rule. We pretrain a Kimi Linear model with 3B activated parameters and 48B total parameters, based on a layerwise hybrid of KDA and Multi-Head Latent Attention (MLA). Our experiments show that with an identical training recipe, Kimi Linear outperforms full MLA with a sizeable margin across all evaluated tasks, while reducing KV cache usage by up to 75% and achieving up to 6 times decoding throughput for a 1M context. These results demonstrate that Kimi Linear can be a drop-in replacement for full attention architectures with superior performance and efficiency, including tasks with longer input and output lengths. To support further research, we open-source the KDA kernel and vLLM implementations, and release the pre-trained and instruction-tuned model checkpoints.
Kimi-VL Technical Report
Apr 10, 2025

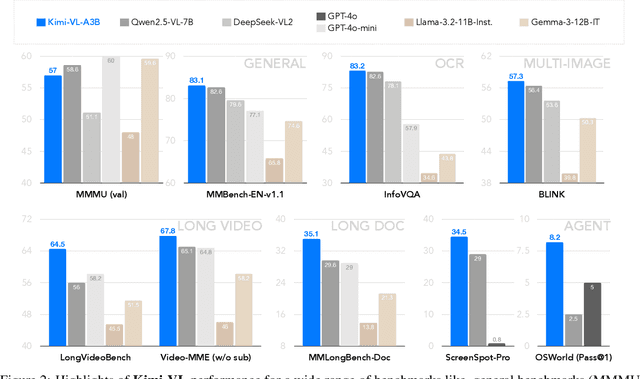
We present Kimi-VL, an efficient open-source Mixture-of-Experts (MoE) vision-language model (VLM) that offers advanced multimodal reasoning, long-context understanding, and strong agent capabilities - all while activating only 2.8B parameters in its language decoder (Kimi-VL-A3B). Kimi-VL demonstrates strong performance across challenging domains: as a general-purpose VLM, Kimi-VL excels in multi-turn agent tasks (e.g., OSWorld), matching flagship models. Furthermore, it exhibits remarkable capabilities across diverse challenging vision language tasks, including college-level image and video comprehension, OCR, mathematical reasoning, and multi-image understanding. In comparative evaluations, it effectively competes with cutting-edge efficient VLMs such as GPT-4o-mini, Qwen2.5-VL-7B, and Gemma-3-12B-IT, while surpassing GPT-4o in several key domains. Kimi-VL also advances in processing long contexts and perceiving clearly. With a 128K extended context window, Kimi-VL can process diverse long inputs, achieving impressive scores of 64.5 on LongVideoBench and 35.1 on MMLongBench-Doc. Its native-resolution vision encoder, MoonViT, further allows it to see and understand ultra-high-resolution visual inputs, achieving 83.2 on InfoVQA and 34.5 on ScreenSpot-Pro, while maintaining lower computational cost for common tasks. Building upon Kimi-VL, we introduce an advanced long-thinking variant: Kimi-VL-Thinking. Developed through long chain-of-thought (CoT) supervised fine-tuning (SFT) and reinforcement learning (RL), this model exhibits strong long-horizon reasoning capabilities. It achieves scores of 61.7 on MMMU, 36.8 on MathVision, and 71.3 on MathVista while maintaining the compact 2.8B activated LLM parameters, setting a new standard for efficient multimodal thinking models. Code and models are publicly accessible at https://github.com/MoonshotAI/Kimi-VL.
MoBA: Mixture of Block Attention for Long-Context LLMs
Feb 18, 2025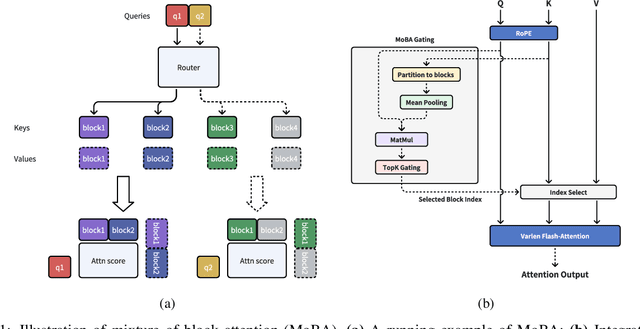
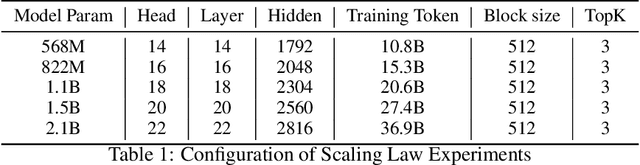
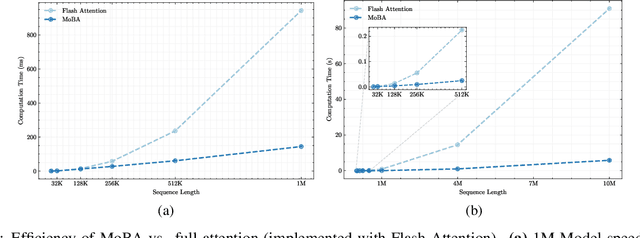
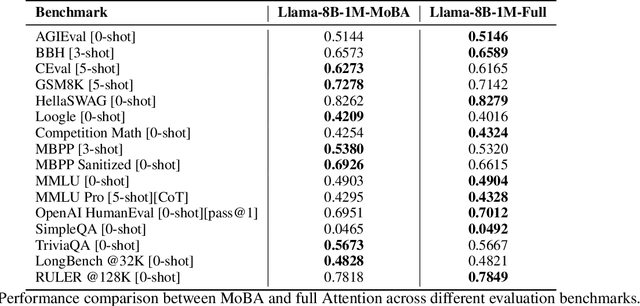
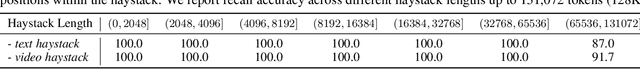
Scaling the effective context length is essential for advancing large language models (LLMs) toward artificial general intelligence (AGI). However, the quadratic increase in computational complexity inherent in traditional attention mechanisms presents a prohibitive overhead. Existing approaches either impose strongly biased structures, such as sink or window attention which are task-specific, or radically modify the attention mechanism into linear approximations, whose performance in complex reasoning tasks remains inadequately explored. In this work, we propose a solution that adheres to the ``less structure'' principle, allowing the model to determine where to attend autonomously, rather than introducing predefined biases. We introduce Mixture of Block Attention (MoBA), an innovative approach that applies the principles of Mixture of Experts (MoE) to the attention mechanism. This novel architecture demonstrates superior performance on long-context tasks while offering a key advantage: the ability to seamlessly transition between full and sparse attention, enhancing efficiency without the risk of compromising performance. MoBA has already been deployed to support Kimi's long-context requests and demonstrates significant advancements in efficient attention computation for LLMs. Our code is available at https://github.com/MoonshotAI/MoBA.
An Autonomous Large Language Model Agent for Chemical Literature Data Mining
Feb 20, 2024



Chemical synthesis, which is crucial for advancing material synthesis and drug discovery, impacts various sectors including environmental science and healthcare. The rise of technology in chemistry has generated extensive chemical data, challenging researchers to discern patterns and refine synthesis processes. Artificial intelligence (AI) helps by analyzing data to optimize synthesis and increase yields. However, AI faces challenges in processing literature data due to the unstructured format and diverse writing style of chemical literature. To overcome these difficulties, we introduce an end-to-end AI agent framework capable of high-fidelity extraction from extensive chemical literature. This AI agent employs large language models (LLMs) for prompt generation and iterative optimization. It functions as a chemistry assistant, automating data collection and analysis, thereby saving manpower and enhancing performance. Our framework's efficacy is evaluated using accuracy, recall, and F1 score of reaction condition data, and we compared our method with human experts in terms of content correctness and time efficiency. The proposed approach marks a significant advancement in automating chemical literature extraction and demonstrates the potential for AI to revolutionize data management and utilization in chemistry.
Holmes: Towards Distributed Training Across Clusters with Heterogeneous NIC Environment
Dec 11, 2023



Large language models (LLMs) such as GPT-3, OPT, and LLaMA have demonstrated remarkable accuracy in a wide range of tasks. However, training these models can incur significant expenses, often requiring tens of thousands of GPUs for months of continuous operation. Typically, this training is carried out in specialized GPU clusters equipped with homogeneous high-speed Remote Direct Memory Access (RDMA) network interface cards (NICs). The acquisition and maintenance of such dedicated clusters is challenging. Current LLM training frameworks, like Megatron-LM and Megatron-DeepSpeed, focus primarily on optimizing training within homogeneous cluster settings. In this paper, we introduce Holmes, a training framework for LLMs that employs thoughtfully crafted data and model parallelism strategies over the heterogeneous NIC environment. Our primary technical contribution lies in a novel scheduling method that intelligently allocates distinct computational tasklets in LLM training to specific groups of GPU devices based on the characteristics of their connected NICs. Furthermore, our proposed framework, utilizing pipeline parallel techniques, demonstrates scalability to multiple GPU clusters, even in scenarios without high-speed interconnects between nodes in distinct clusters. We conducted comprehensive experiments that involved various scenarios in the heterogeneous NIC environment. In most cases, our framework achieves performance levels close to those achievable with homogeneous RDMA-capable networks (InfiniBand or RoCE), significantly exceeding training efficiency within the pure Ethernet environment. Additionally, we verified that our framework outperforms other mainstream LLM frameworks under heterogeneous NIC environment in terms of training efficiency and can be seamlessly integrated with them.
Towards an Automatic AI Agent for Reaction Condition Recommendation in Chemical Synthesis
Nov 28, 2023



Artificial intelligence (AI) for reaction condition optimization has become an important topic in the pharmaceutical industry, given that a data-driven AI model can assist drug discovery and accelerate reaction design. However, existing AI models lack the chemical insights and real-time knowledge acquisition abilities of experienced human chemists. This paper proposes a Large Language Model (LLM) empowered AI agent to bridge this gap. We put forth a novel three-phase paradigm and applied advanced intelligence-enhancement methods like in-context learning and multi-LLM debate so that the AI agent can borrow human insight and update its knowledge by searching the latest chemical literature. Additionally, we introduce a novel Coarse-label Contrastive Learning (CCL) based chemical fingerprint that greatly enhances the agent's performance in optimizing the reaction condition. With the above efforts, the proposed AI agent can autonomously generate the optimal reaction condition recommendation without any human interaction. Further, the agent is highly professional in terms of chemical reactions. It demonstrates close-to-human performance and strong generalization capability in both dry-lab and wet-lab experiments. As the first attempt in the chemical AI agent, this work goes a step further in the field of "AI for chemistry" and opens up new possibilities for computer-aided synthesis planning.
Multi-task Bioassay Pre-training for Protein-ligand Binding Affinity Prediction
Jun 08, 2023



Protein-ligand binding affinity (PLBA) prediction is the fundamental task in drug discovery. Recently, various deep learning-based models predict binding affinity by incorporating the three-dimensional structure of protein-ligand complexes as input and achieving astounding progress. However, due to the scarcity of high-quality training data, the generalization ability of current models is still limited. In addition, different bioassays use varying affinity measurement labels (i.e., IC50, Ki, Kd), and different experimental conditions inevitably introduce systematic noise, which poses a significant challenge to constructing high-precision affinity prediction models. To address these issues, we (1) propose Multi-task Bioassay Pre-training (MBP), a pre-training framework for structure-based PLBA prediction; (2) construct a pre-training dataset called ChEMBL-Dock with more than 300k experimentally measured affinity labels and about 2.8M docked three-dimensional structures. By introducing multi-task pre-training to treat the prediction of different affinity labels as different tasks and classifying relative rankings between samples from the same bioassay, MBP learns robust and transferrable structural knowledge from our new ChEMBL-Dock dataset with varied and noisy labels. Experiments substantiate the capability of MBP as a general framework that can improve and be tailored to mainstream structure-based PLBA prediction tasks. To the best of our knowledge, MBP is the first affinity pre-training model and shows great potential for future development.
Multi-Constraint Molecular Generation using Sparsely Labelled Training Data for Localized High-Concentration Electrolyte Diluent Screening
Jan 12, 2023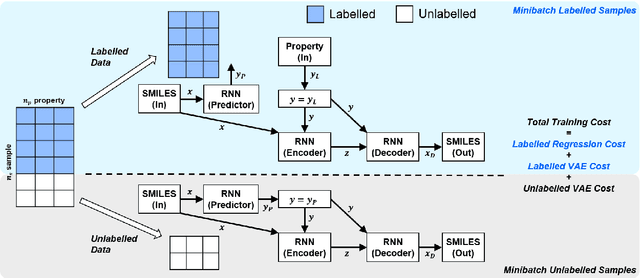
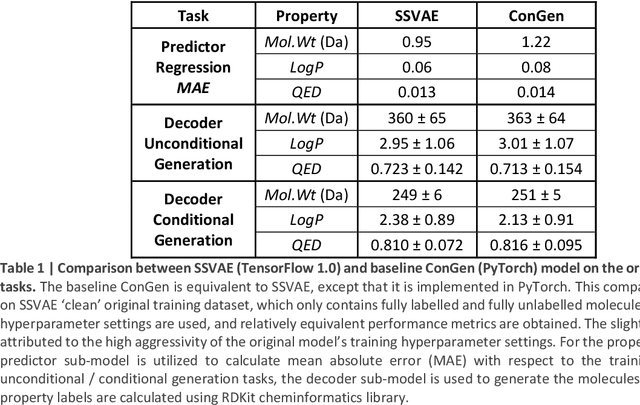
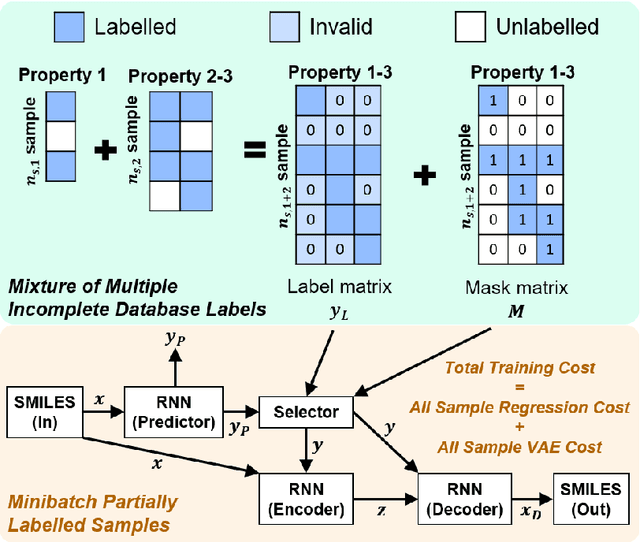
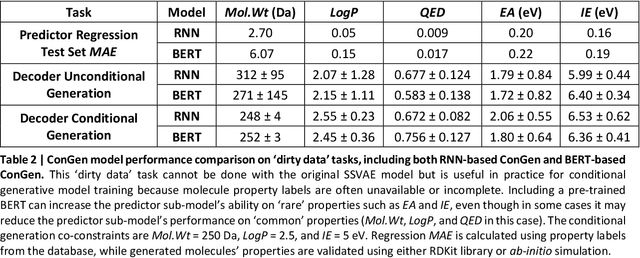
Recently, machine learning methods have been used to propose molecules with desired properties, which is especially useful for exploring large chemical spaces efficiently. However, these methods rely on fully labelled training data, and are not practical in situations where molecules with multiple property constraints are required. There is often insufficient training data for all those properties from publicly available databases, especially when ab-initio simulation or experimental property data is also desired for training the conditional molecular generative model. In this work, we show how to modify a semi-supervised variational auto-encoder (SSVAE) model which only works with fully labelled and fully unlabelled molecular property training data into the ConGen model, which also works on training data that have sparsely populated labels. We evaluate ConGen's performance in generating molecules with multiple constraints when trained on a dataset combined from multiple publicly available molecule property databases, and demonstrate an example application of building the virtual chemical space for potential Lithium-ion battery localized high-concentration electrolyte (LHCE) diluents.
Protein-Ligand Complex Generator & Drug Screening via Tiered Tensor Transform
Jan 03, 2023



Accurate determination of a small molecule candidate (ligand) binding pose in its target protein pocket is important for computer-aided drug discovery. Typical rigid-body docking methods ignore the pocket flexibility of protein, while the more accurate pose generation using molecular dynamics is hindered by slow protein dynamics. We develop a tiered tensor transform (3T) algorithm to rapidly generate diverse protein-ligand complex conformations for both pose and affinity estimation in drug screening, requiring neither machine learning training nor lengthy dynamics computation, while maintaining both coarse-grain-like coordinated protein dynamics and atomistic-level details of the complex pocket. The 3T conformation structures we generate are closer to experimental co-crystal structures than those generated by docking software, and more importantly achieve significantly higher accuracy in active ligand classification than traditional ensemble docking using hundreds of experimental protein conformations. 3T structure transformation is decoupled from the system physics, making future usage in other computational scientific domains possible.