 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSaDiT: Efficient Protein Backbone Design via Latent Structural Tokenization and Diffusion Transformers
Feb 06, 2026Generative models for de novo protein backbone design have achieved remarkable success in creating novel protein structures. However, these diffusion-based approaches remain computationally intensive and slower than desired for large-scale structural exploration. While recent efforts like Proteina have introduced flow-matching to improve sampling efficiency, the potential of tokenization for structural compression and acceleration remains largely unexplored in the protein domain. In this work, we present SaDiT, a novel framework that accelerates protein backbone generation by integrating SaProt Tokenization with a Diffusion Transformer (DiT) architecture. SaDiT leverages a discrete latent space to represent protein geometry, significantly reducing the complexity of the generation process while maintaining theoretical SE(3) equivalence. To further enhance efficiency, we introduce an IPA Token Cache mechanism that optimizes the Invariant Point Attention (IPA) layers by reusing computed token states during iterative sampling. Experimental results demonstrate that SaDiT outperforms state-of-the-art models, including RFDiffusion and Proteina, in both computational speed and structural viability. We evaluate our model across unconditional backbone generation and fold-class conditional generation tasks, where SaDiT shows superior ability to capture complex topological features with high designability.
Boosting RL-Based Visual Reasoning with Selective Adversarial Entropy Intervention
Dec 11, 2025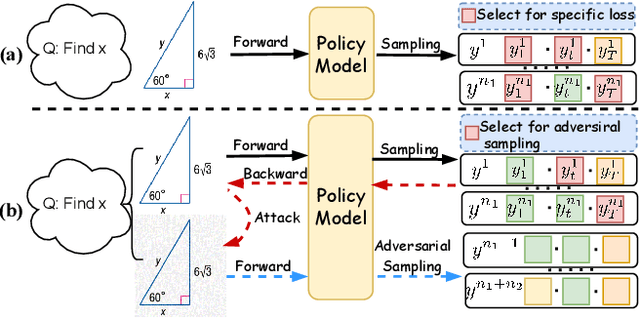
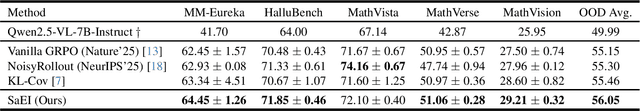
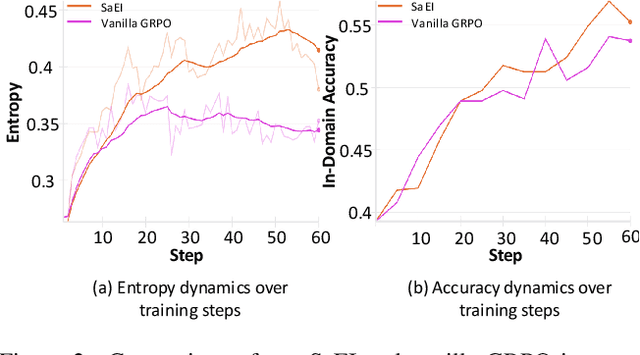
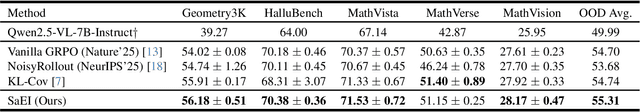
Recently, reinforcement learning (RL) has become a common choice in enhancing the reasoning capabilities of vision-language models (VLMs). Considering existing RL-based finetuning methods, entropy intervention turns out to be an effective way to benefit exploratory ability, thereby improving policy performance. Notably, most existing studies intervene in entropy by simply controlling the update of specific tokens during policy optimization of RL. They ignore the entropy intervention during the RL sampling that can boost the performance of GRPO by improving the diversity of responses. In this paper, we propose Selective-adversarial Entropy Intervention, namely SaEI, which enhances policy entropy by distorting the visual input with the token-selective adversarial objective coming from the entropy of sampled responses. Specifically, we first propose entropy-guided adversarial sampling (EgAS) that formulates the entropy of sampled responses as an adversarial objective. Then, the corresponding adversarial gradient can be used to attack the visual input for producing adversarial samples, allowing the policy model to explore a larger answer space during RL sampling. Then, we propose token-selective entropy computation (TsEC) to maximize the effectiveness of adversarial attack in EgAS without distorting factual knowledge within VLMs. Extensive experiments on both in-domain and out-of-domain datasets show that our proposed method can greatly improve policy exploration via entropy intervention, to boost reasoning capabilities. Code will be released once the paper is accepted.
Scrutinize What We Ignore: Reining Task Representation Shift In Context-Based Offline Meta Reinforcement Learning
May 20, 2024



Offline meta reinforcement learning (OMRL) has emerged as a promising approach for interaction avoidance and strong generalization performance by leveraging pre-collected data and meta-learning techniques. Previous context-based approaches predominantly rely on the intuition that maximizing the mutual information between the task and the task representation ($I(Z;M)$) can lead to performance improvements. Despite achieving attractive results, the theoretical justification of performance improvement for such intuition has been lacking. Motivated by the return discrepancy scheme in the model-based RL field, we find that maximizing $I(Z;M)$ can be interpreted as consistently raising the lower bound of the expected return for a given policy conditioning on the optimal task representation. However, this optimization process ignores the task representation shift between two consecutive updates, which may lead to performance improvement collapse. To address this problem, we turn to use the framework of performance difference bound to consider the impacts of task representation shift explicitly. We demonstrate that by reining the task representation shift, it is possible to achieve monotonic performance improvements, thereby showcasing the advantage against previous approaches. To make it practical, we design an easy yet highly effective algorithm RETRO (\underline{RE}ining \underline{T}ask \underline{R}epresentation shift in context-based \underline{O}ffline meta reinforcement learning) with only adding one line of code compared to the backbone. Empirical results validate its state-of-the-art (SOTA) asymptotic performance, training stability and training-time consumption on MuJoCo and MetaWorld benchmarks.
An Autonomous Large Language Model Agent for Chemical Literature Data Mining
Feb 20, 2024



Chemical synthesis, which is crucial for advancing material synthesis and drug discovery, impacts various sectors including environmental science and healthcare. The rise of technology in chemistry has generated extensive chemical data, challenging researchers to discern patterns and refine synthesis processes. Artificial intelligence (AI) helps by analyzing data to optimize synthesis and increase yields. However, AI faces challenges in processing literature data due to the unstructured format and diverse writing style of chemical literature. To overcome these difficulties, we introduce an end-to-end AI agent framework capable of high-fidelity extraction from extensive chemical literature. This AI agent employs large language models (LLMs) for prompt generation and iterative optimization. It functions as a chemistry assistant, automating data collection and analysis, thereby saving manpower and enhancing performance. Our framework's efficacy is evaluated using accuracy, recall, and F1 score of reaction condition data, and we compared our method with human experts in terms of content correctness and time efficiency. The proposed approach marks a significant advancement in automating chemical literature extraction and demonstrates the potential for AI to revolutionize data management and utilization in chemistry.
Towards an Information Theoretic Framework of Context-Based Offline Meta-Reinforcement Learning
Feb 04, 2024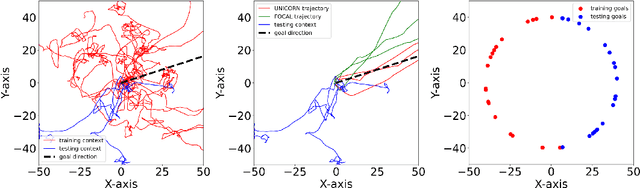
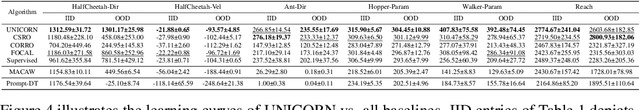
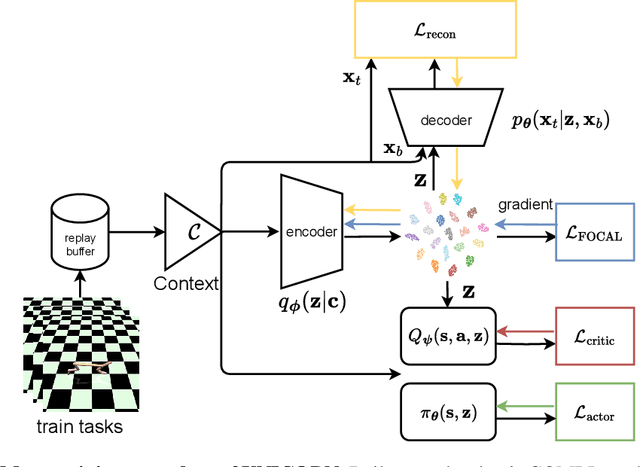
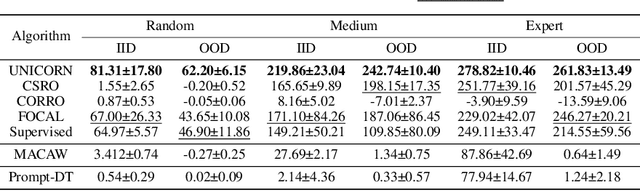
As a marriage between offline RL and meta-RL, the advent of offline meta-reinforcement learning (OMRL) has shown great promise in enabling RL agents to multi-task and quickly adapt while acquiring knowledge safely. Among which, Context-based OMRL (COMRL) as a popular paradigm, aims to learn a universal policy conditioned on effective task representations. In this work, by examining several key milestones in the field of COMRL, we propose to integrate these seemingly independent methodologies into a unified information theoretic framework. Most importantly, we show that the pre-existing COMRL algorithms are essentially optimizing the same mutual information objective between the task variable $\boldsymbol{M}$ and its latent representation $\boldsymbol{Z}$ by implementing various approximate bounds. Based on the theoretical insight and the information bottleneck principle, we arrive at a novel algorithm dubbed UNICORN, which exhibits remarkable generalization across a broad spectrum of RL benchmarks, context shift scenarios, data qualities and deep learning architectures, attaining the new state-of-the-art. We believe that our framework could open up avenues for new optimality bounds and COMRL algorithms.
Towards an Automatic AI Agent for Reaction Condition Recommendation in Chemical Synthesis
Nov 28, 2023



Artificial intelligence (AI) for reaction condition optimization has become an important topic in the pharmaceutical industry, given that a data-driven AI model can assist drug discovery and accelerate reaction design. However, existing AI models lack the chemical insights and real-time knowledge acquisition abilities of experienced human chemists. This paper proposes a Large Language Model (LLM) empowered AI agent to bridge this gap. We put forth a novel three-phase paradigm and applied advanced intelligence-enhancement methods like in-context learning and multi-LLM debate so that the AI agent can borrow human insight and update its knowledge by searching the latest chemical literature. Additionally, we introduce a novel Coarse-label Contrastive Learning (CCL) based chemical fingerprint that greatly enhances the agent's performance in optimizing the reaction condition. With the above efforts, the proposed AI agent can autonomously generate the optimal reaction condition recommendation without any human interaction. Further, the agent is highly professional in terms of chemical reactions. It demonstrates close-to-human performance and strong generalization capability in both dry-lab and wet-lab experiments. As the first attempt in the chemical AI agent, this work goes a step further in the field of "AI for chemistry" and opens up new possibilities for computer-aided synthesis planning.
MolKD: Distilling Cross-Modal Knowledge in Chemical Reactions for Molecular Property Prediction
May 03, 2023How to effectively represent molecules is a long-standing challenge for molecular property prediction and drug discovery. This paper studies this problem and proposes to incorporate chemical domain knowledge, specifically related to chemical reactions, for learning effective molecular representations. However, the inherent cross-modality property between chemical reactions and molecules presents a significant challenge to address. To this end, we introduce a novel method, namely MolKD, which Distills cross-modal Knowledge in chemical reactions to assist Molecular property prediction. Specifically, the reaction-to-molecule distillation model within MolKD transfers cross-modal knowledge from a pre-trained teacher network learning with one modality (i.e., reactions) into a student network learning with another modality (i.e., molecules). Moreover, MolKD learns effective molecular representations by incorporating reaction yields to measure transformation efficiency of the reactant-product pair when pre-training on reactions. Extensive experiments demonstrate that MolKD significantly outperforms various competitive baseline models, e.g., 2.1% absolute AUC-ROC gain on Tox21. Further investigations demonstrate that pre-trained molecular representations in MolKD can distinguish chemically reasonable molecular similarities, which enables molecular property prediction with high robustness and interpretability.
Reweighted Mixup for Subpopulation Shift
Apr 09, 2023Subpopulation shift exists widely in many real-world applications, which refers to the training and test distributions that contain the same subpopulation groups but with different subpopulation proportions. Ignoring subpopulation shifts may lead to significant performance degradation and fairness concerns. Importance reweighting is a classical and effective way to handle the subpopulation shift. However, recent studies have recognized that most of these approaches fail to improve the performance especially when applied to over-parameterized neural networks which are capable of fitting any training samples. In this work, we propose a simple yet practical framework, called reweighted mixup (RMIX), to mitigate the overfitting issue in over-parameterized models by conducting importance weighting on the ''mixed'' samples. Benefiting from leveraging reweighting in mixup, RMIX allows the model to explore the vicinal space of minority samples more, thereby obtaining more robust model against subpopulation shift. When the subpopulation memberships are unknown, the training-trajectories-based uncertainty estimation is equipped in the proposed RMIX to flexibly characterize the subpopulation distribution. We also provide insightful theoretical analysis to verify that RMIX achieves better generalization bounds over prior works. Further, we conduct extensive empirical studies across a wide range of tasks to validate the effectiveness of the proposed method.
Deploying Offline Reinforcement Learning with Human Feedback
Mar 13, 2023
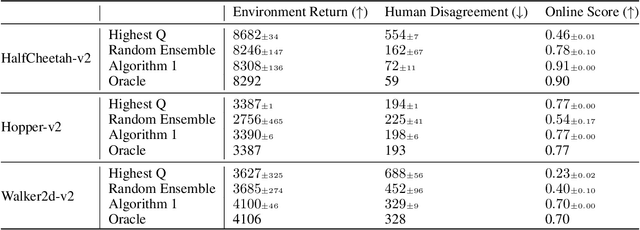


Reinforcement learning (RL) has shown promise for decision-making tasks in real-world applications. One practical framework involves training parameterized policy models from an offline dataset and subsequently deploying them in an online environment. However, this approach can be risky since the offline training may not be perfect, leading to poor performance of the RL models that may take dangerous actions. To address this issue, we propose an alternative framework that involves a human supervising the RL models and providing additional feedback in the online deployment phase. We formalize this online deployment problem and develop two approaches. The first approach uses model selection and the upper confidence bound algorithm to adaptively select a model to deploy from a candidate set of trained offline RL models. The second approach involves fine-tuning the model in the online deployment phase when a supervision signal arrives. We demonstrate the effectiveness of these approaches for robot locomotion control and traffic light control tasks through empirical validation.
Handling Missing Data via Max-Entropy Regularized Graph Autoencoder
Nov 30, 2022



Graph neural networks (GNNs) are popular weapons for modeling relational data. Existing GNNs are not specified for attribute-incomplete graphs, making missing attribute imputation a burning issue. Until recently, many works notice that GNNs are coupled with spectral concentration, which means the spectrum obtained by GNNs concentrates on a local part in spectral domain, e.g., low-frequency due to oversmoothing issue. As a consequence, GNNs may be seriously flawed for reconstructing graph attributes as graph spectral concentration tends to cause a low imputation precision. In this work, we present a regularized graph autoencoder for graph attribute imputation, named MEGAE, which aims at mitigating spectral concentration problem by maximizing the graph spectral entropy. Notably, we first present the method for estimating graph spectral entropy without the eigen-decomposition of Laplacian matrix and provide the theoretical upper error bound. A maximum entropy regularization then acts in the latent space, which directly increases the graph spectral entropy. Extensive experiments show that MEGAE outperforms all the other state-of-the-art imputation methods on a variety of benchmark datasets.