 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Universal Density Matrix Functional from Molecular Orbital-Based Machine Learning: Transferability across Organic Molecules
Paper and Code
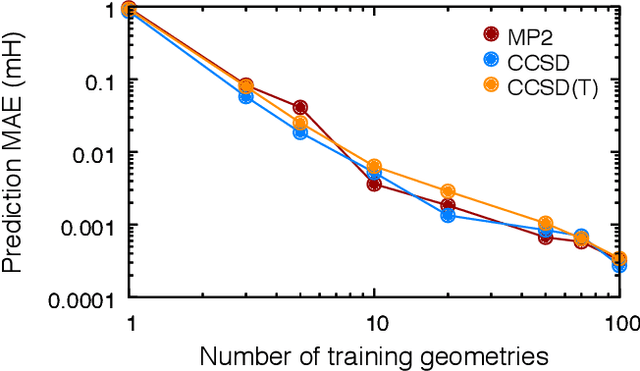
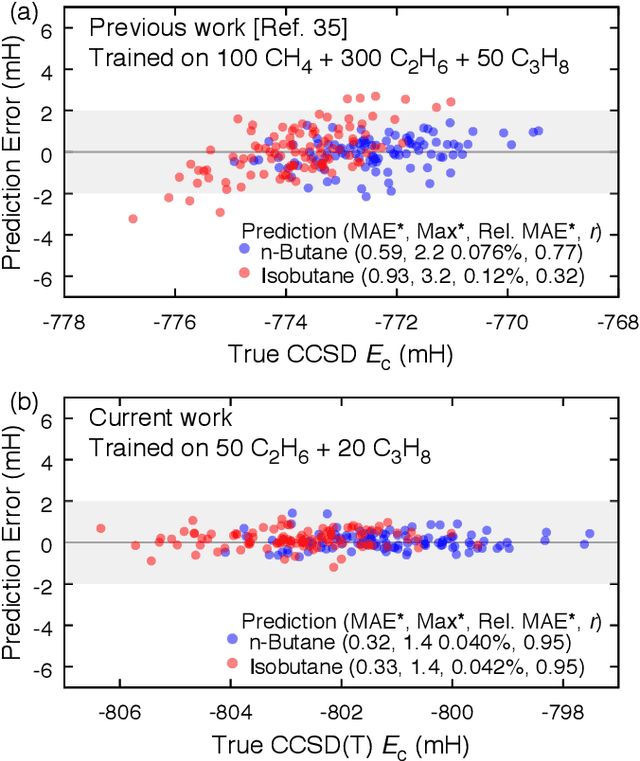
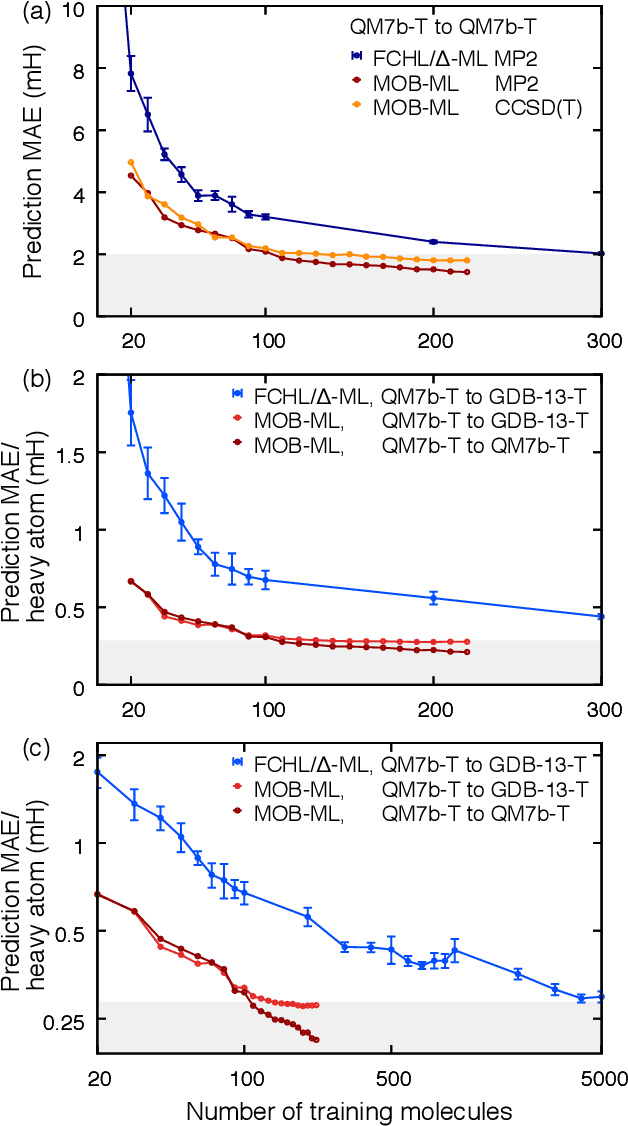
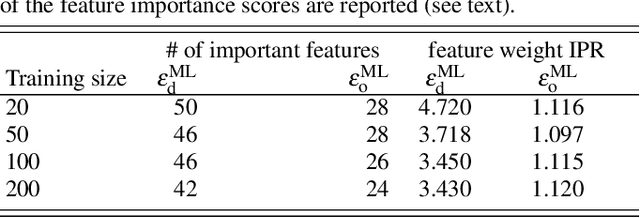
We address the degree to which machine learning can be used to accurately and transferably predict post-Hartree-Fock correlation energies. After presenting refined strategies for feature design and selection, the molecular-orbital-based machine learning (MOB-ML) method is first applied to benchmark test systems. It is shown that the total electronic energy for a set of 1000 randomized geometries of water can be described to within 1 millihartree using a model that is trained at the level of MP2, CCSD, or CCSD(T) using only a single reference calculation at a randomized geometry. To explore the breadth of chemical diversity that can be described, the MOB-ML method is then applied a set of 7211 organic models with up to seven heavy atoms. It is shown that MP2 calculations on only 90 molecules are needed to train a model that predicts MP2 energies to within 2 millihartree accuracy for the remaining 7121 molecules; likewise, CCSD(T) calculations on only 150 molecules are needed to train a model that predicts CCSD(T) energies for the remaining molecules to within 2 millihartree accuracy. The MP2 model, trained with only 90 reference calculations on seven-heavy-atom molecules, is then applied to a diverse set of 1000 thirteen-heavy-atom organic molecules, demonstrating transferable preservation of chemical accuracy.