 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePILOT: Equivariant diffusion for pocket conditioned de novo ligand generation with multi-objective guidance via importance sampling
May 23, 2024



The generation of ligands that both are tailored to a given protein pocket and exhibit a range of desired chemical properties is a major challenge in structure-based drug design. Here, we propose an in-silico approach for the $\textit{de novo}$ generation of 3D ligand structures using the equivariant diffusion model PILOT, combining pocket conditioning with a large-scale pre-training and property guidance. Its multi-objective trajectory-based importance sampling strategy is designed to direct the model towards molecules that not only exhibit desired characteristics such as increased binding affinity for a given protein pocket but also maintains high synthetic accessibility. This ensures the practicality of sampled molecules, thus maximizing their potential for the drug discovery pipeline. PILOT significantly outperforms existing methods across various metrics on the common benchmark dataset CrossDocked2020. Moreover, we employ PILOT to generate novel ligands for unseen protein pockets from the Kinodata-3D dataset, which encompasses a substantial portion of the human kinome. The generated structures exhibit predicted $IC_{50}$ values indicative of potent biological activity, which highlights the potential of PILOT as a powerful tool for structure-based drug design.
SchNetPack 2.0: A neural network toolbox for atomistic machine learning
Dec 11, 2022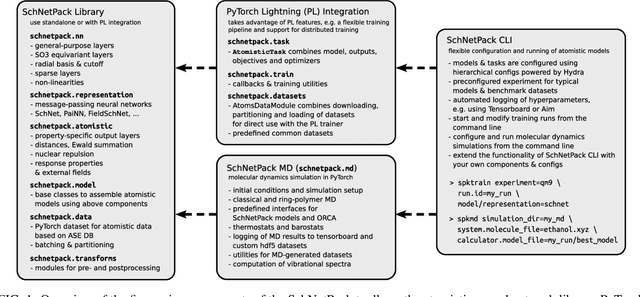
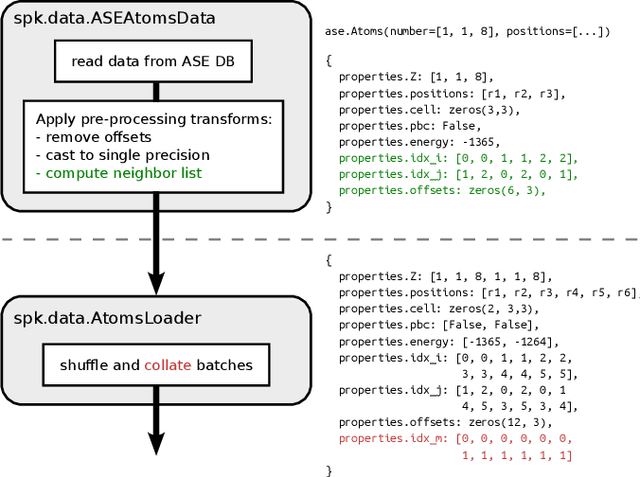
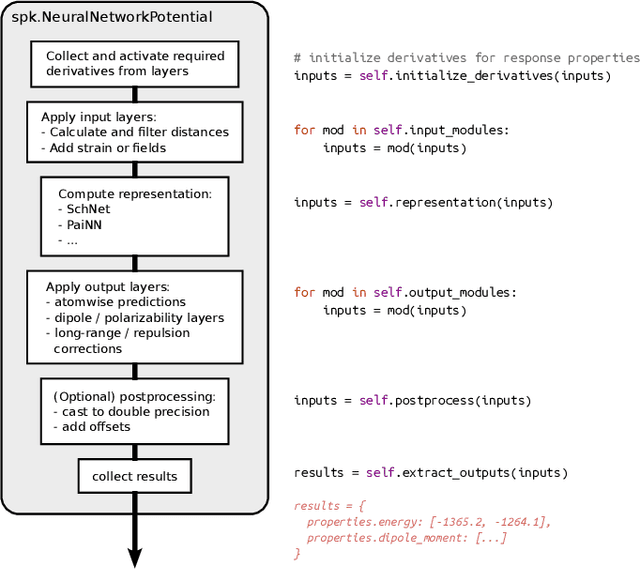

SchNetPack is a versatile neural networks toolbox that addresses both the requirements of method development and application of atomistic machine learning. Version 2.0 comes with an improved data pipeline, modules for equivariant neural networks as well as a PyTorch implementation of molecular dynamics. An optional integration with PyTorch Lightning and the Hydra configuration framework powers a flexible command-line interface. This makes SchNetPack 2.0 easily extendable with custom code and ready for complex training task such as generation of 3d molecular structures.
Automatic Identification of Chemical Moieties
Mar 30, 2022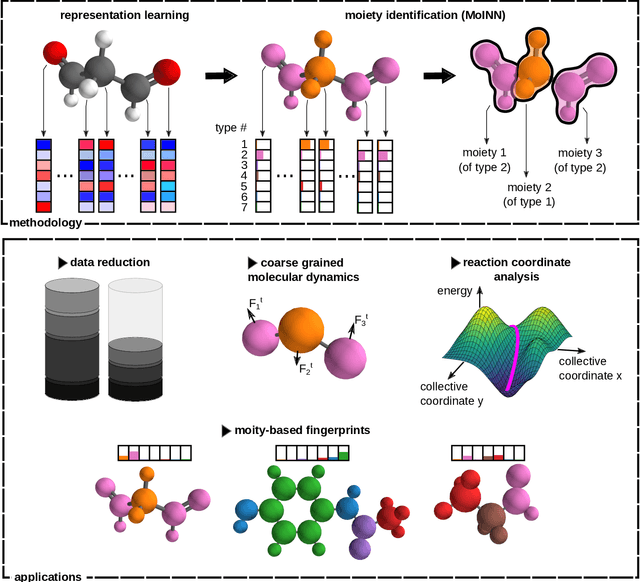
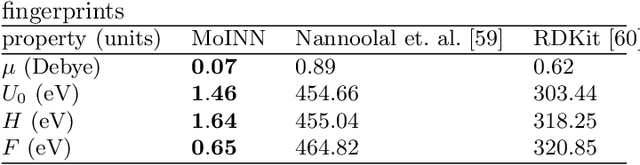
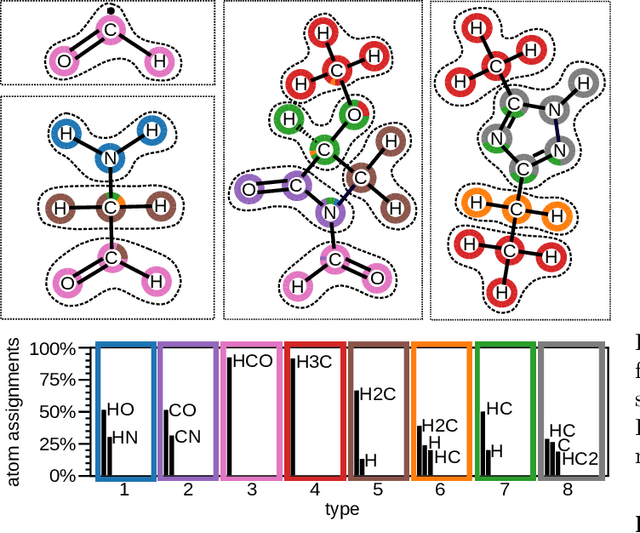
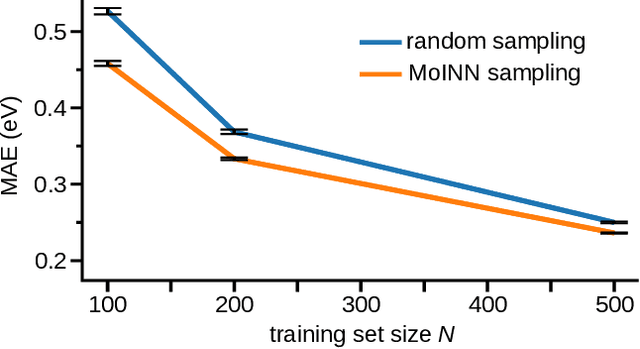
In recent years, the prediction of quantum mechanical observables with machine learning methods has become increasingly popular. Message-passing neural networks (MPNNs) solve this task by constructing atomic representations, from which the properties of interest are predicted. Here, we introduce a method to automatically identify chemical moieties (molecular building blocks) from such representations, enabling a variety of applications beyond property prediction, which otherwise rely on expert knowledge. The required representation can either be provided by a pretrained MPNN, or learned from scratch using only structural information. Beyond the data-driven design of molecular fingerprints, the versatility of our approach is demonstrated by enabling the selection of representative entries in chemical databases, the automatic construction of coarse-grained force fields, as well as the identification of reaction coordinates.
Inverse design of 3d molecular structures with conditional generative neural networks
Sep 10, 2021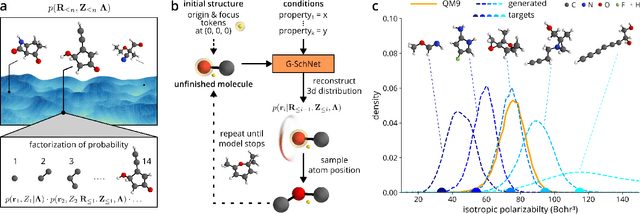
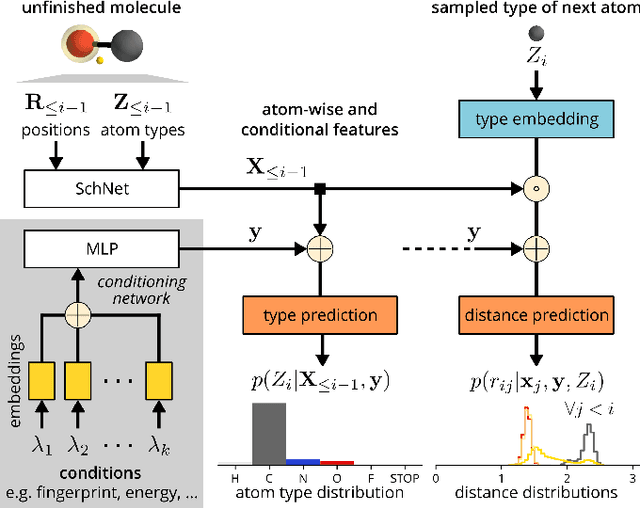
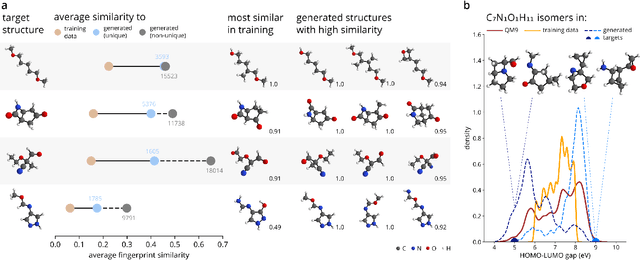
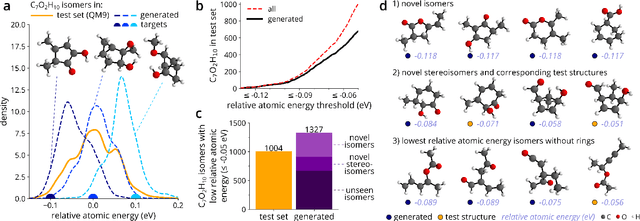
The rational design of molecules with desired properties is a long-standing challenge in chemistry. Generative neural networks have emerged as a powerful approach to sample novel molecules from a learned distribution. Here, we propose a conditional generative neural network for 3d molecular structures with specified structural and chemical properties. This approach is agnostic to chemical bonding and enables targeted sampling of novel molecules from conditional distributions, even in domains where reference calculations are sparse. We demonstrate the utility of our method for inverse design by generating molecules with specified composition or motifs, discovering particularly stable molecules, and jointly targeting multiple electronic properties beyond the training regime.
SpookyNet: Learning Force Fields with Electronic Degrees of Freedom and Nonlocal Effects
May 01, 2021



In recent years, machine-learned force fields (ML-FFs) have gained increasing popularity in the field of computational chemistry. Provided they are trained on appropriate reference data, ML-FFs combine the accuracy of ab initio methods with the efficiency of conventional force fields. However, current ML-FFs typically ignore electronic degrees of freedom, such as the total charge or spin, when forming their prediction. In addition, they often assume chemical locality, which can be problematic in cases where nonlocal effects play a significant role. This work introduces SpookyNet, a deep neural network for constructing ML-FFs with explicit treatment of electronic degrees of freedom and quantum nonlocality. Its predictions are further augmented with physically-motivated corrections to improve the description of long-ranged interactions and nuclear repulsion. SpookyNet improves upon the current state-of-the-art (or achieves similar performance) on popular quantum chemistry data sets. Notably, it can leverage the learned chemical insights, e.g. by predicting unknown spin states or by properly modeling physical limits. Moreover, it is able to generalize across chemical and conformational space and thus close an important remaining gap for today's machine learning models in quantum chemistry.
Equivariant message passing for the prediction of tensorial properties and molecular spectra
Feb 08, 2021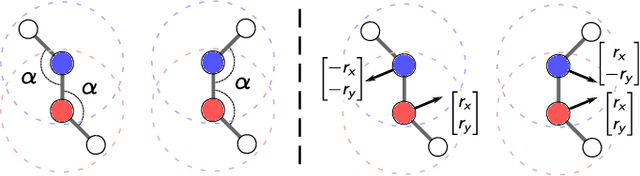
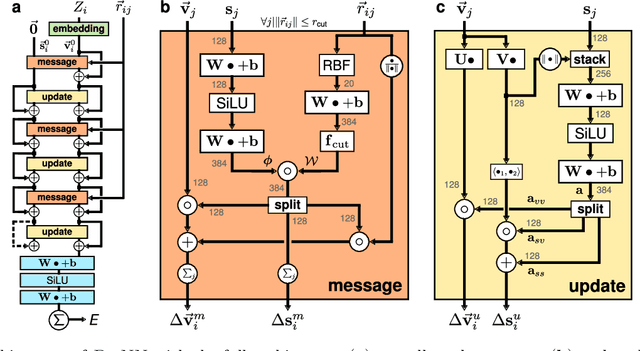
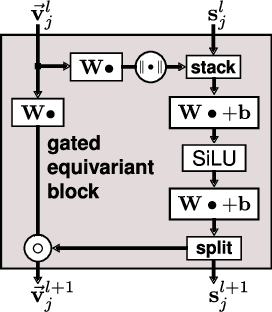
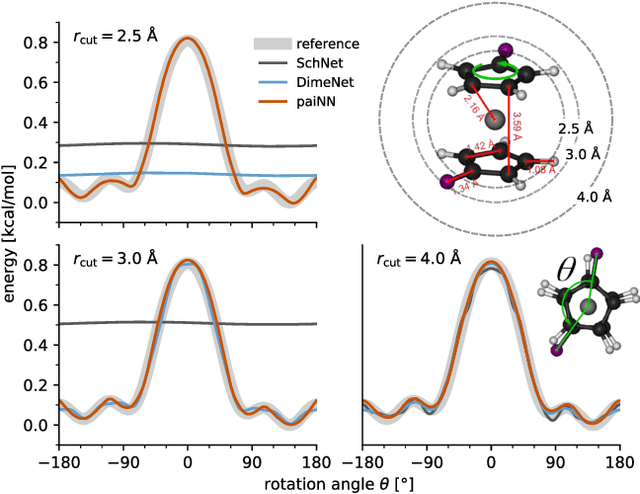
Message passing neural networks have become a method of choice for learning on graphs, in particular the prediction of chemical properties and the acceleration of molecular dynamics studies. While they readily scale to large training data sets, previous approaches have proven to be less data efficient than kernel methods. We identify limitations of invariant representations as a major reason and extend the message passing formulation to rotationally equivariant representations. On this basis, we propose the polarizable atom interaction neural network (PaiNN) and improve on common molecule benchmarks over previous networks, while reducing model size and inference time. We leverage the equivariant atomwise representations obtained by PaiNN for the prediction of tensorial properties. Finally, we apply this to the simulation of molecular spectra, achieving speedups of 4-5 orders of magnitude compared to the electronic structure reference.
Machine learning of solvent effects on molecular spectra and reactions
Nov 04, 2020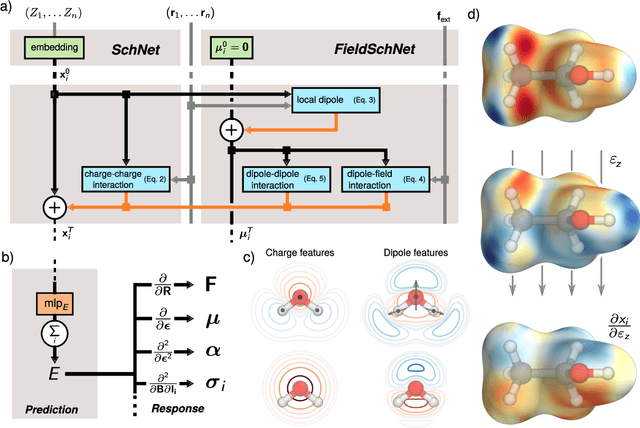
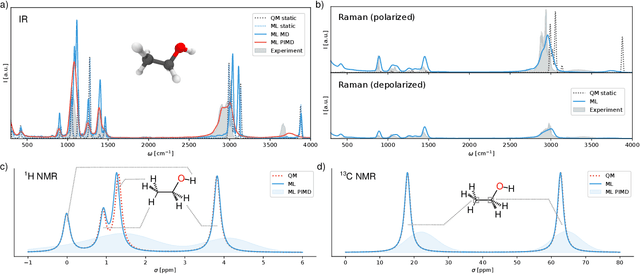
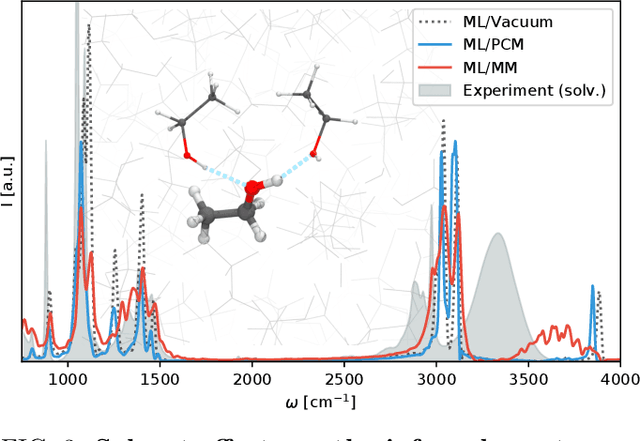
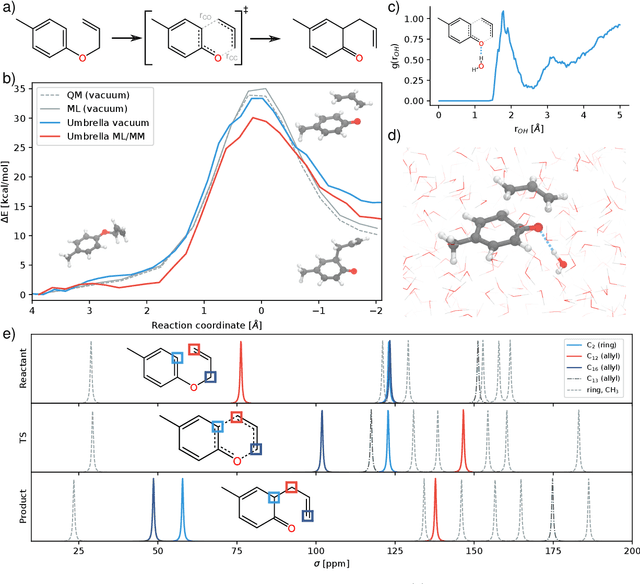
Fast and accurate simulation of complex chemical systems in environments such as solutions is a long standing challenge in theoretical chemistry. In recent years, machine learning has extended the boundaries of quantum chemistry by providing highly accurate and efficient surrogate models of electronic structure theory, which previously have been out of reach for conventional approaches. Those models have long been restricted to closed molecular systems without accounting for environmental influences, such as external electric and magnetic fields or solvent effects. Here, we introduce the deep neural network FieldSchNet for modeling the interaction of molecules with arbitrary external fields. FieldSchNet offers access to a wealth of molecular response properties, enabling it to simulate a wide range of molecular spectra, such as infrared, Raman and nuclear magnetic resonance. Beyond that, it is able to describe implicit and explicit molecular environments, operating as a polarizable continuum model for solvation or in a quantum mechanics / molecular mechanics setup. We employ FieldSchNet to study the influence of solvent effects on molecular spectra and a Claisen rearrangement reaction. Based on these results, we use FieldSchNet to design an external environment capable of lowering the activation barrier of the rearrangement reaction significantly, demonstrating promising venues for inverse chemical design.
Machine Learning Force Fields
Oct 14, 2020
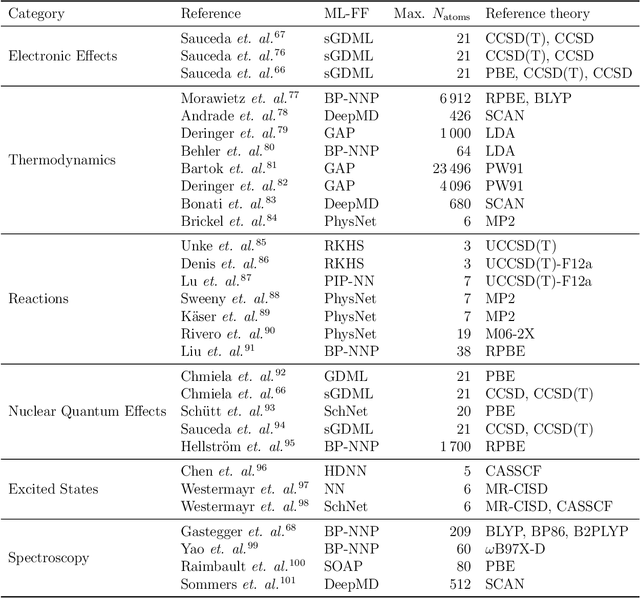
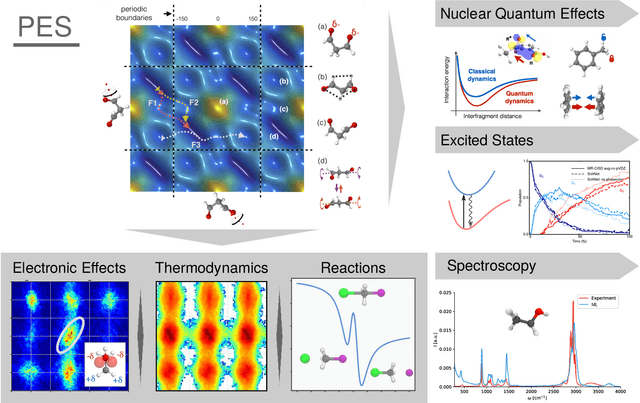

In recent years, the use of Machine Learning (ML) in computational chemistry has enabled numerous advances previously out of reach due to the computational complexity of traditional electronic-structure methods. One of the most promising applications is the construction of ML-based force fields (FFs), with the aim to narrow the gap between the accuracy of ab initio methods and the efficiency of classical FFs. The key idea is to learn the statistical relation between chemical structure and potential energy without relying on a preconceived notion of fixed chemical bonds or knowledge about the relevant interactions. Such universal ML approximations are in principle only limited by the quality and quantity of the reference data used to train them. This review gives an overview of applications of ML-FFs and the chemical insights that can be obtained from them. The core concepts underlying ML-FFs are described in detail and a step-by-step guide for constructing and testing them from scratch is given. The text concludes with a discussion of the challenges that remain to be overcome by the next generation of ML-FFs.
XAI for Graphs: Explaining Graph Neural Network Predictions by Identifying Relevant Walks
Jun 12, 2020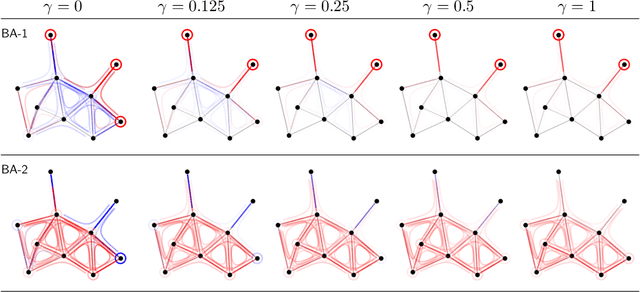
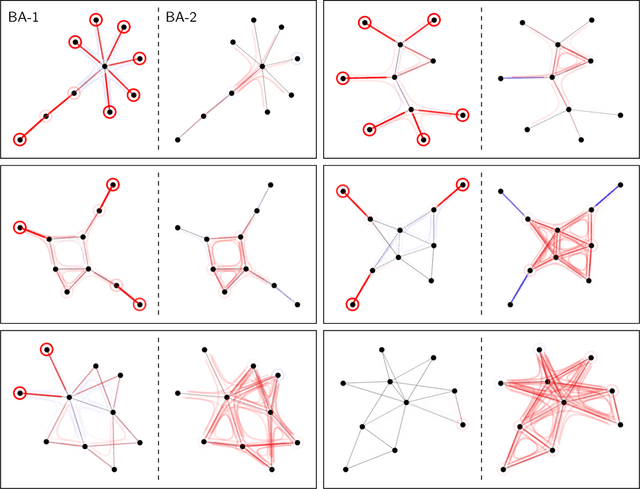
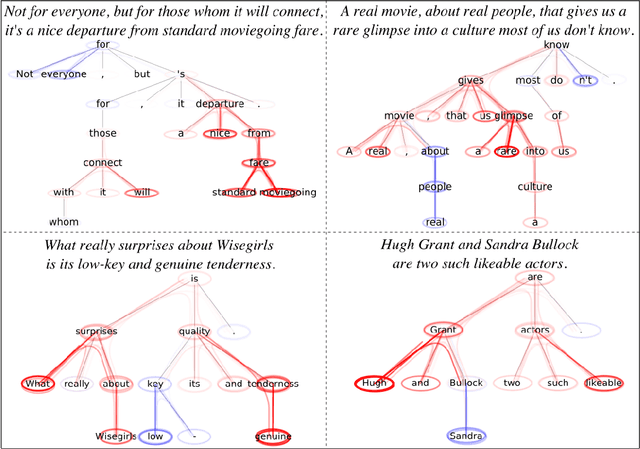
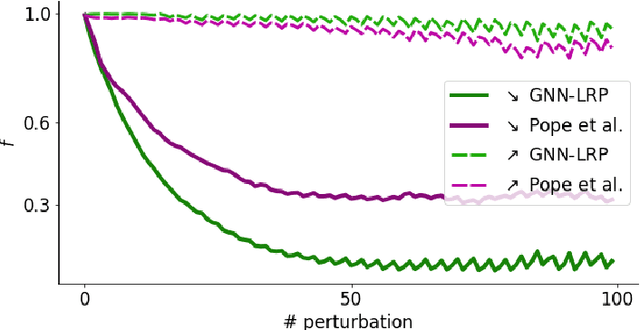
Graph Neural Networks (GNNs) are a popular approach for predicting graph structured data. As GNNs tightly entangle the input graph into the neural network structure, common explainable AI (XAI) approaches are not applicable. To a large extent, GNNs have remained black-boxes for the user so far. In this paper, we contribute by proposing a new XAI approach for GNNs. Our approach is derived from high-order Taylor expansions and is able to generate a decomposition of the GNN prediction as a collection of relevant walks on the input graph. We find that these high-order Taylor expansions can be equivalently (and more simply) computed using multiple backpropagation passes from the top layer of the GNN to the first layer. The explanation can then be further robustified and generalized by using layer-wise-relevance propagation (LRP) in place of the standard equations for gradient propagation. Our novel method which we denote as 'GNN-LRP' is tested on scale-free graphs, sentence parsing trees, molecular graphs, and pixel lattices representing images. In each case, it performs stably and accurately, and delivers interesting and novel application insights.
Autonomous robotic nanofabrication with reinforcement learning
Feb 27, 2020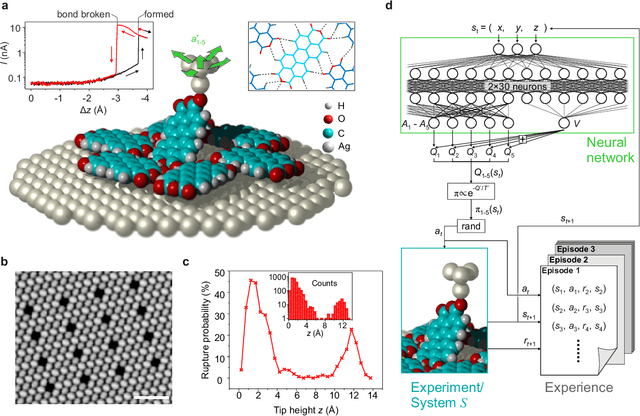
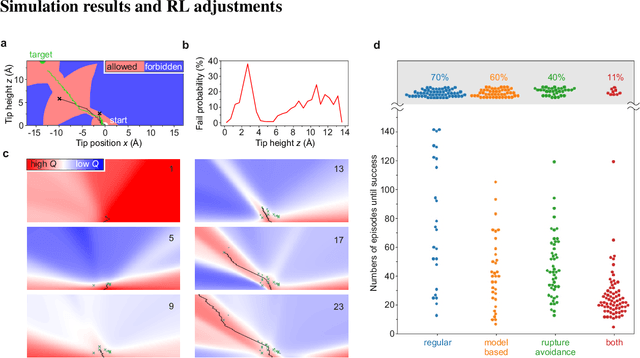
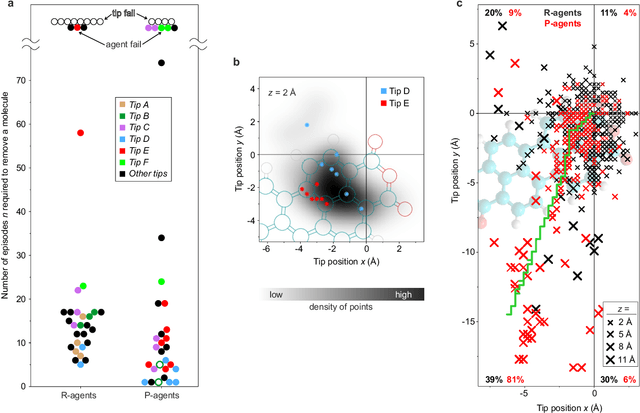
The ability to handle single molecules as effectively as macroscopic building-blocks would enable the construction of complex supramolecular structures that are not accessible by self-assembly. The fundamental challenges on the way towards this goal are the uncontrolled variability and poor observability of atomic-scale conformations. Here, we present a strategy to work around both obstacles, and demonstrate autonomous robotic nanofabrication by manipulating single molecules. Our approach employs reinforcement learning (RL), which is able to learn solution strategies even in the face of large uncertainty and with sparse feedback. However, to be useful for autonomous nanofabrication, standard RL algorithms need to be adapted to cope with the limited training opportunities available. We demonstrate the potential of our RL approach by applying it to an exemplary task of subtractive manufacturing, the removal of individual molecules from a molecular layer using a scanning probe microscope (SPM). Our RL agent reaches an excellent performance level, enabling us to automate a task which previously had to be performed by a human. We anticipate that our work opens the way towards autonomous agents for the robotic construction of functional supramolecular structures with speed, precision and perseverance beyond our current capabilities.