 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA practical guide to machine learning interatomic potentials -- Status and future
Mar 12, 2025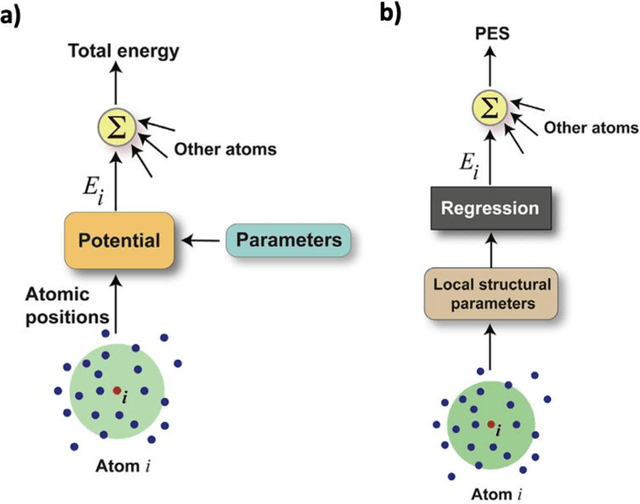

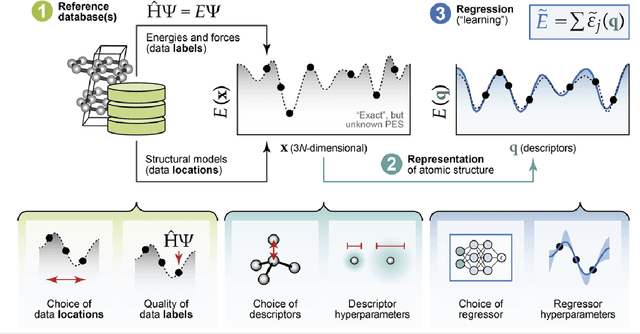
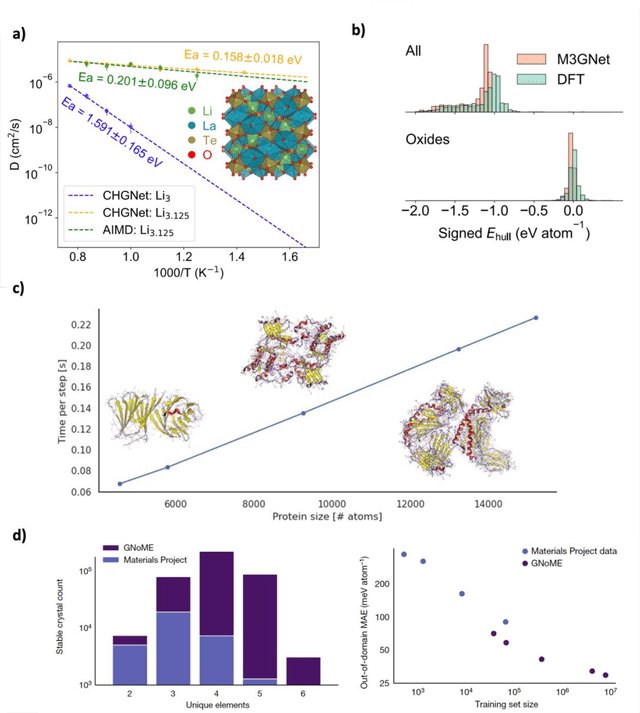
The rapid development and large body of literature on machine learning interatomic potentials (MLIPs) can make it difficult to know how to proceed for researchers who are not experts but wish to use these tools. The spirit of this review is to help such researchers by serving as a practical, accessible guide to the state-of-the-art in MLIPs. This review paper covers a broad range of topics related to MLIPs, including (i) central aspects of how and why MLIPs are enablers of many exciting advancements in molecular modeling, (ii) the main underpinnings of different types of MLIPs, including their basic structure and formalism, (iii) the potentially transformative impact of universal MLIPs for both organic and inorganic systems, including an overview of the most recent advances, capabilities, downsides, and potential applications of this nascent class of MLIPs, (iv) a practical guide for estimating and understanding the execution speed of MLIPs, including guidance for users based on hardware availability, type of MLIP used, and prospective simulation size and time, (v) a manual for what MLIP a user should choose for a given application by considering hardware resources, speed requirements, energy and force accuracy requirements, as well as guidance for choosing pre-trained potentials or fitting a new potential from scratch, (vi) discussion around MLIP infrastructure, including sources of training data, pre-trained potentials, and hardware resources for training, (vii) summary of some key limitations of present MLIPs and current approaches to mitigate such limitations, including methods of including long-range interactions, handling magnetic systems, and treatment of excited states, and finally (viii) we finish with some more speculative thoughts on what the future holds for the development and application of MLIPs over the next 3-10+ years.
Machine Learning Force Fields
Oct 14, 2020
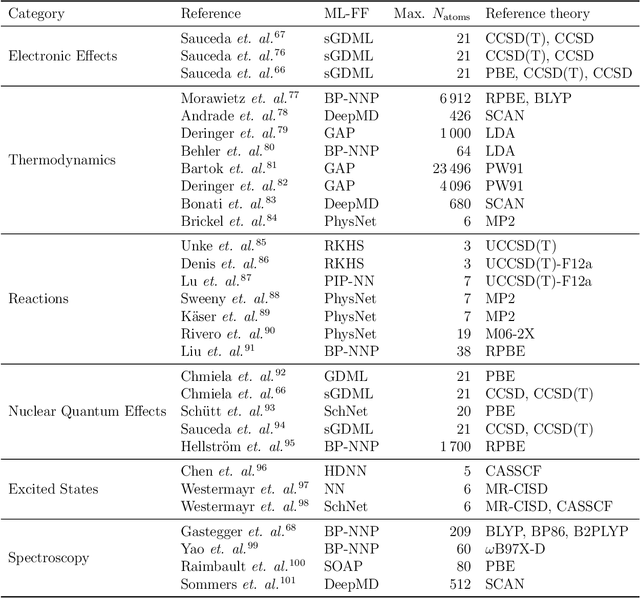
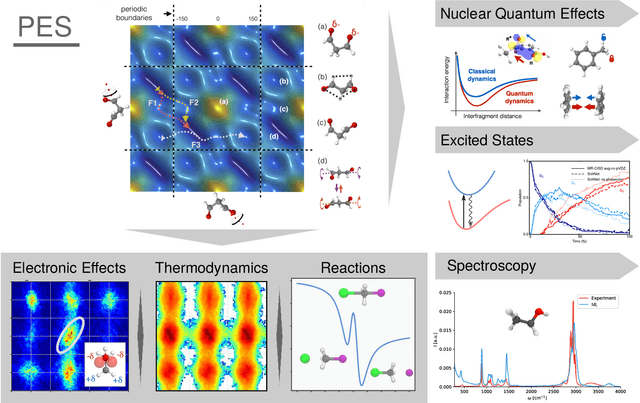

In recent years, the use of Machine Learning (ML) in computational chemistry has enabled numerous advances previously out of reach due to the computational complexity of traditional electronic-structure methods. One of the most promising applications is the construction of ML-based force fields (FFs), with the aim to narrow the gap between the accuracy of ab initio methods and the efficiency of classical FFs. The key idea is to learn the statistical relation between chemical structure and potential energy without relying on a preconceived notion of fixed chemical bonds or knowledge about the relevant interactions. Such universal ML approximations are in principle only limited by the quality and quantity of the reference data used to train them. This review gives an overview of applications of ML-FFs and the chemical insights that can be obtained from them. The core concepts underlying ML-FFs are described in detail and a step-by-step guide for constructing and testing them from scratch is given. The text concludes with a discussion of the challenges that remain to be overcome by the next generation of ML-FFs.