 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCharacterizing High-Capacity Janus Aminobenzene-Graphene Anode for Sodium-Ion Batteries with Machine Learning
Mar 23, 2026Sodium-ion batteries require anodes that combine high capacity, low operating voltage, fast Na-ion transport, and mechanical stability, which conventional anodes struggle to deliver. Here, we use the SpookyNet machine-learning force field (MLFF) together with all-electron density-functional theory calculations to characterize Na storage in aminobenzene-functionalized Janus graphene (Na$_x$AB) at room-temperature. Simulations across state of charge reveal a three-stage storage mechanism-site-specific adsorption at aminobenzene groups and Na$_n$@AB$_m$ structure formation, followed by interlayer gallery filling-contrasting the multi-stage pore-, graphite-interlayer-, and defect-controlled behavior in hard carbon. This leads to an OCV profile with an extended low-voltage plateau of 0.15 V vs. Na/Na$^{+}$, an estimated gravimetric capacity of $\sim$400 mAh g$^{-1}$, negligible volume change, and Na diffusivities of $\sim10^{-6}$ cm$^{2}$ s$^{-1}$, two to three orders of magnitude higher than in hard carbon. Our results establish Janus aminobenzene-graphene as a promising, structurally defined high-capacity Na-ion anode and illustrate the power of MLFF-based simulations for characterizing electrode materials.
Machine Learned Force Fields: Fundamentals, its reach, and challenges
Mar 07, 2025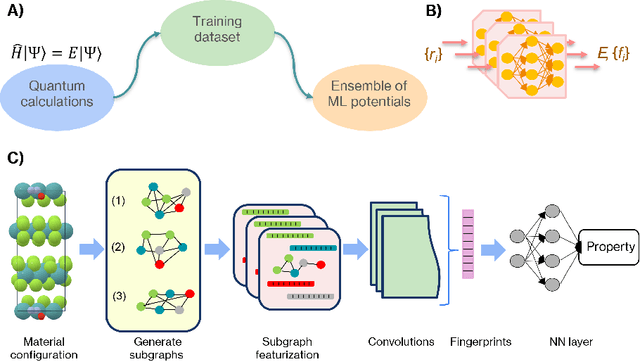

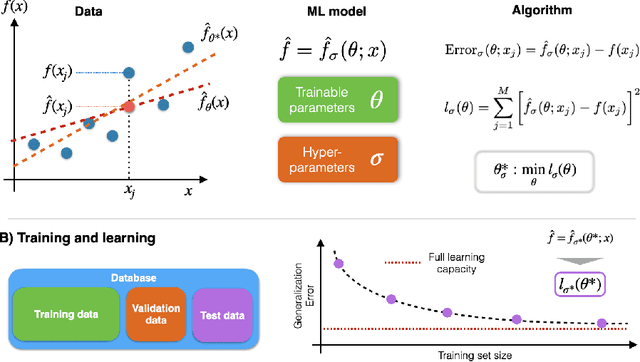
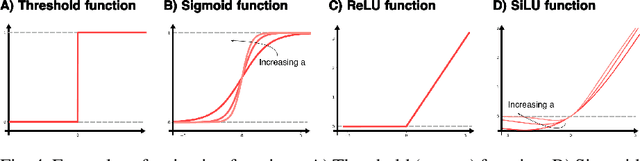
Highly accurate force fields are a mandatory requirement to generate predictive simulations. In this regard, Machine Learning Force Fields (MLFFs) have emerged as a revolutionary approach in computational chemistry and materials science, combining the accuracy of quantum mechanical methods with computational efficiency orders of magnitude superior to ab-initio methods. This chapter provides an introduction of the fundamentals of learning and how it is applied to construct MLFFs, detailing key methodologies such as neural network potentials and kernel-based models. Emphasis is placed on the construction of SchNet model, as one of the most elemental neural network-based force fields that are nowadays the basis of modern architectures. Additionally, the GDML framework is described in detail as an example of how the elegant formulation of kernel methods can be used to construct mathematically robust and physics-inspired MLFFs. The ongoing advancements in MLFF development continue to expand their applicability, enabling precise simulations of large and complex systems that were previously beyond reach. This chapter concludes by highlighting the transformative impact of MLFFs on scientific research, underscoring their role in driving future discoveries in the fields of chemistry, physics, and materials science.
Super-resolution in Molecular Dynamics Trajectory Reconstruction with Bi-Directional Neural Networks
Jan 02, 2022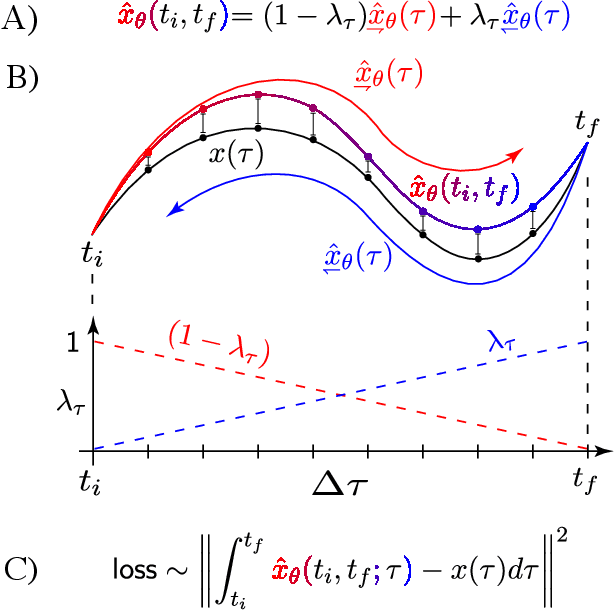
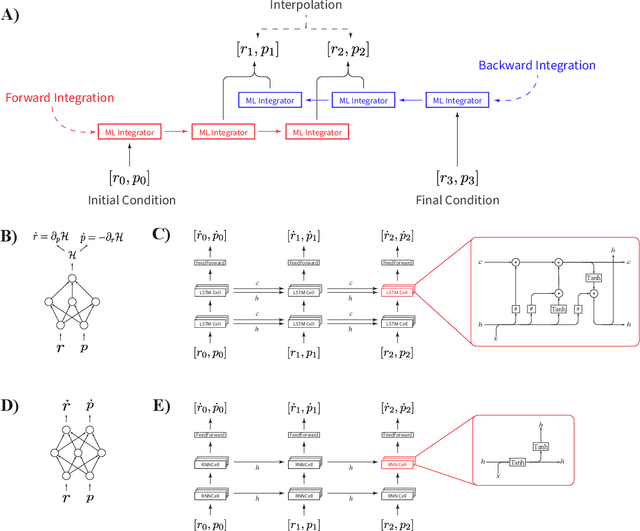
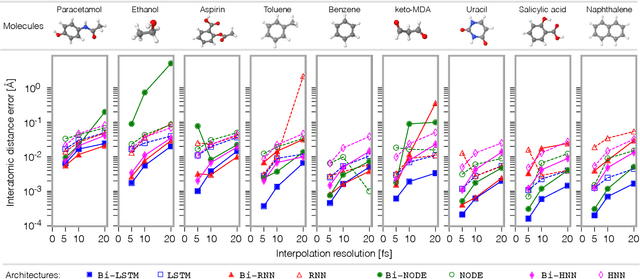
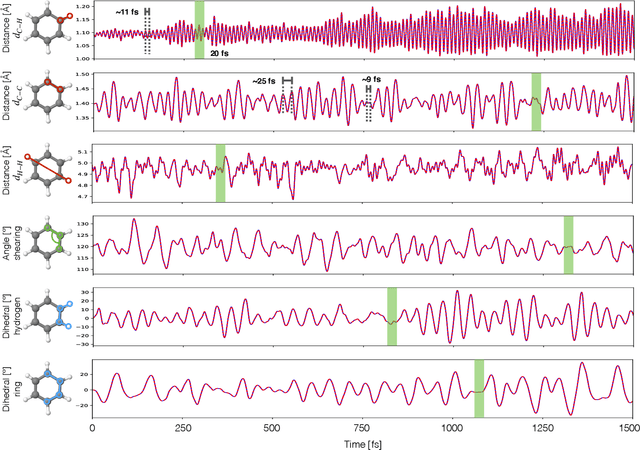
Molecular dynamics simulations are a cornerstone in science, allowing to investigate from the system's thermodynamics to analyse intricate molecular interactions. In general, to create extended molecular trajectories can be a computationally expensive process, for example, when running $ab-initio$ simulations. Hence, repeating such calculations to either obtain more accurate thermodynamics or to get a higher resolution in the dynamics generated by a fine-grained quantum interaction can be time- and computationally-consuming. In this work, we explore different machine learning (ML) methodologies to increase the resolution of molecular dynamics trajectories on-demand within a post-processing step. As a proof of concept, we analyse the performance of bi-directional neural networks such as neural ODEs, Hamiltonian networks, recurrent neural networks and LSTMs, as well as the uni-directional variants as a reference, for molecular dynamics simulations (here: the MD17 dataset). We have found that Bi-LSTMs are the best performing models; by utilizing the local time-symmetry of thermostated trajectories they can even learn long-range correlations and display high robustness to noisy dynamics across molecular complexity. Our models can reach accuracies of up to 10$^{-4}$ angstroms in trajectory interpolation, while faithfully reconstructing several full cycles of unseen intricate high-frequency molecular vibrations, rendering the comparison between the learned and reference trajectories indistinguishable. The results reported in this work can serve (1) as a baseline for larger systems, as well as (2) for the construction of better MD integrators.
BIGDML: Towards Exact Machine Learning Force Fields for Materials
Jun 08, 2021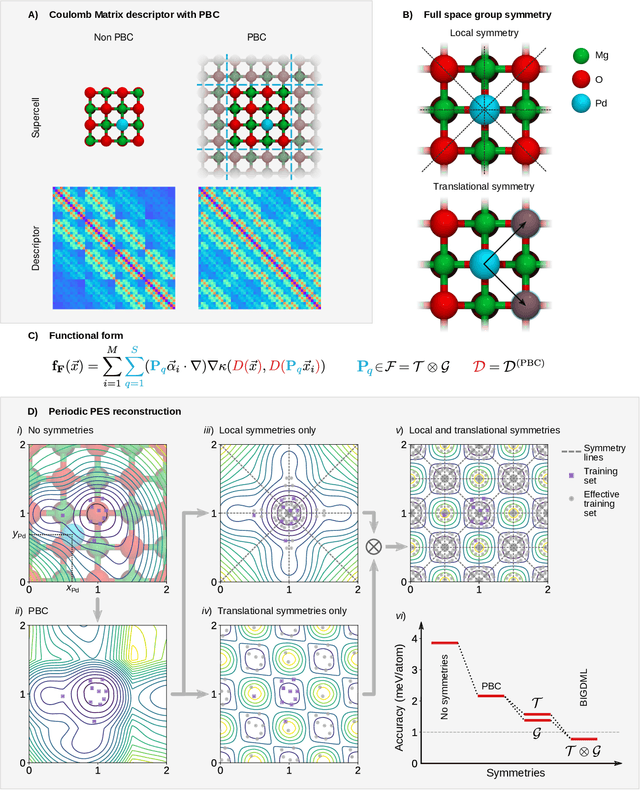
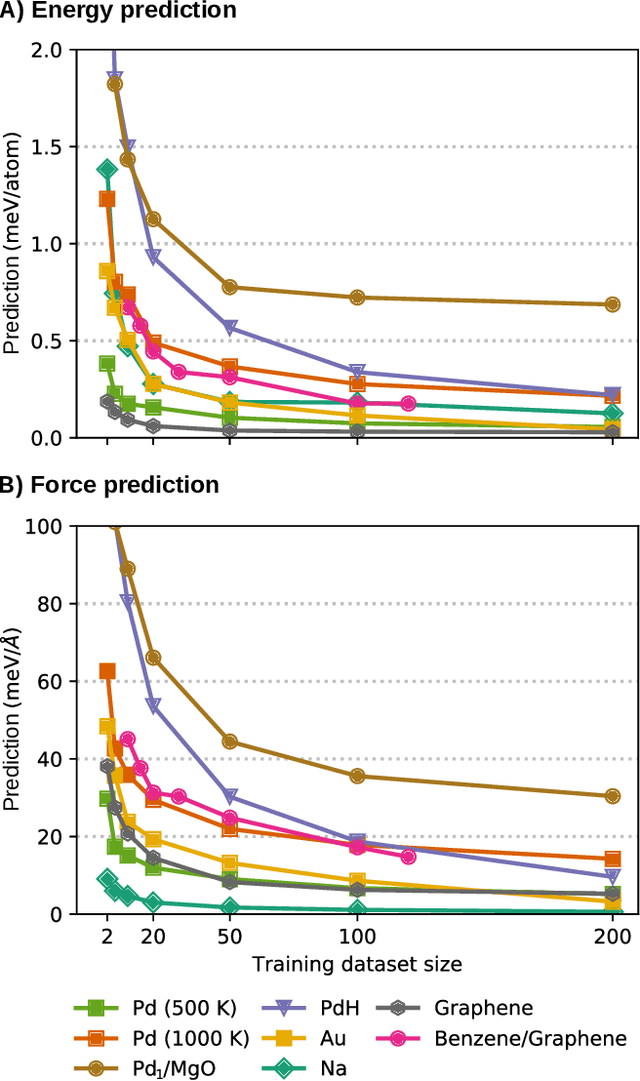
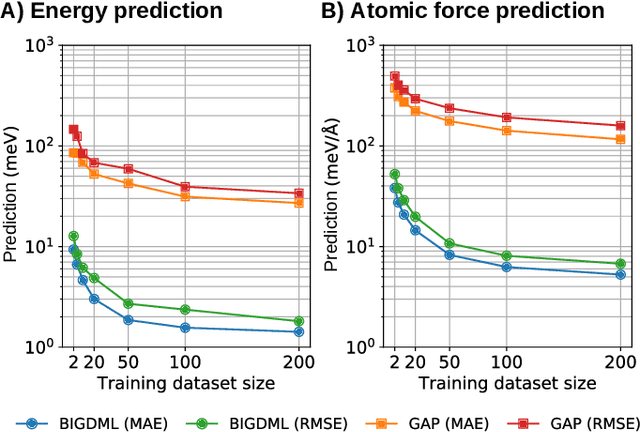
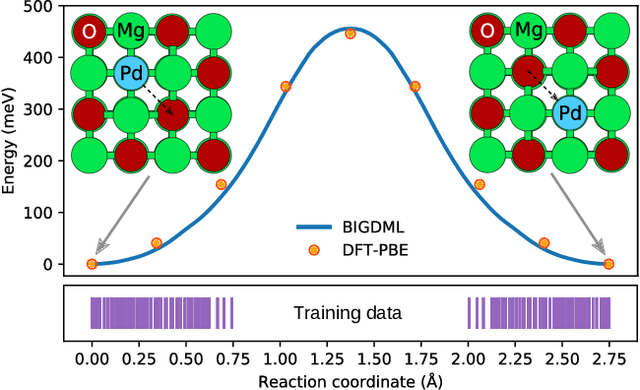
Machine-learning force fields (MLFF) should be accurate, computationally and data efficient, and applicable to molecules, materials, and interfaces thereof. Currently, MLFFs often introduce tradeoffs that restrict their practical applicability to small subsets of chemical space or require exhaustive datasets for training. Here, we introduce the Bravais-Inspired Gradient-Domain Machine Learning (BIGDML) approach and demonstrate its ability to construct reliable force fields using a training set with just 10-200 geometries for materials including pristine and defect-containing 2D and 3D semiconductors and metals, as well as chemisorbed and physisorbed atomic and molecular adsorbates on surfaces. The BIGDML model employs the full relevant symmetry group for a given material, does not assume artificial atom types or localization of atomic interactions and exhibits high data efficiency and state-of-the-art energy accuracies (errors substantially below 1 meV per atom) for an extended set of materials. Extensive path-integral molecular dynamics carried out with BIGDML models demonstrate the counterintuitive localization of benzene--graphene dynamics induced by nuclear quantum effects and allow to rationalize the Arrhenius behavior of hydrogen diffusion coefficient in a Pd crystal for a wide range of temperatures.
SpookyNet: Learning Force Fields with Electronic Degrees of Freedom and Nonlocal Effects
May 01, 2021



In recent years, machine-learned force fields (ML-FFs) have gained increasing popularity in the field of computational chemistry. Provided they are trained on appropriate reference data, ML-FFs combine the accuracy of ab initio methods with the efficiency of conventional force fields. However, current ML-FFs typically ignore electronic degrees of freedom, such as the total charge or spin, when forming their prediction. In addition, they often assume chemical locality, which can be problematic in cases where nonlocal effects play a significant role. This work introduces SpookyNet, a deep neural network for constructing ML-FFs with explicit treatment of electronic degrees of freedom and quantum nonlocality. Its predictions are further augmented with physically-motivated corrections to improve the description of long-ranged interactions and nuclear repulsion. SpookyNet improves upon the current state-of-the-art (or achieves similar performance) on popular quantum chemistry data sets. Notably, it can leverage the learned chemical insights, e.g. by predicting unknown spin states or by properly modeling physical limits. Moreover, it is able to generalize across chemical and conformational space and thus close an important remaining gap for today's machine learning models in quantum chemistry.
Machine Learning Force Fields
Oct 14, 2020
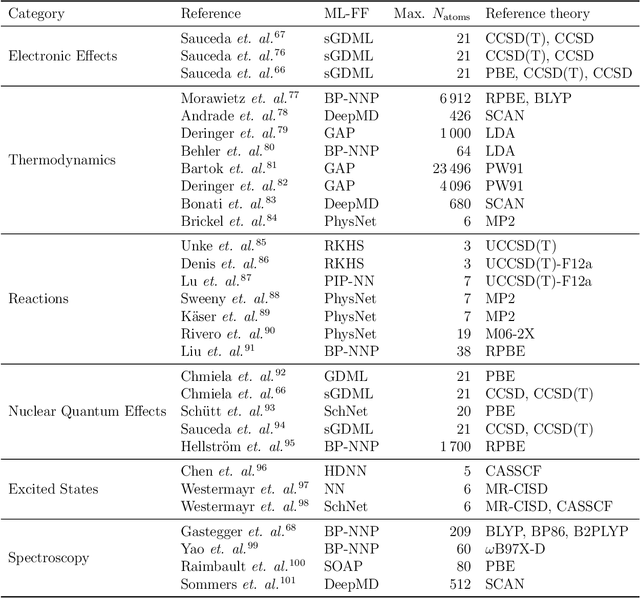
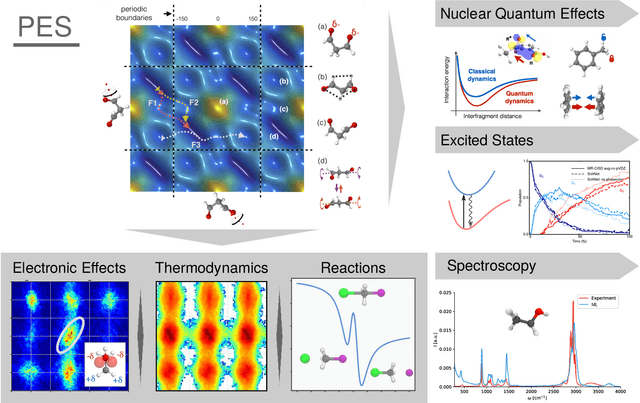

In recent years, the use of Machine Learning (ML) in computational chemistry has enabled numerous advances previously out of reach due to the computational complexity of traditional electronic-structure methods. One of the most promising applications is the construction of ML-based force fields (FFs), with the aim to narrow the gap between the accuracy of ab initio methods and the efficiency of classical FFs. The key idea is to learn the statistical relation between chemical structure and potential energy without relying on a preconceived notion of fixed chemical bonds or knowledge about the relevant interactions. Such universal ML approximations are in principle only limited by the quality and quantity of the reference data used to train them. This review gives an overview of applications of ML-FFs and the chemical insights that can be obtained from them. The core concepts underlying ML-FFs are described in detail and a step-by-step guide for constructing and testing them from scratch is given. The text concludes with a discussion of the challenges that remain to be overcome by the next generation of ML-FFs.
SchNet: A continuous-filter convolutional neural network for modeling quantum interactions
Dec 19, 2017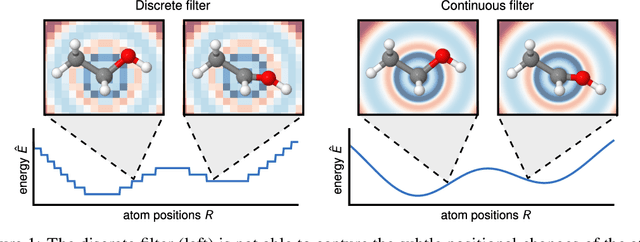

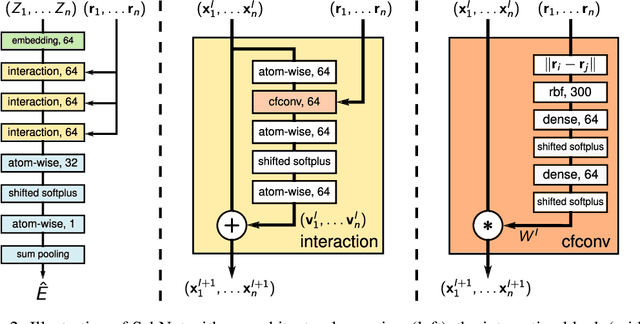
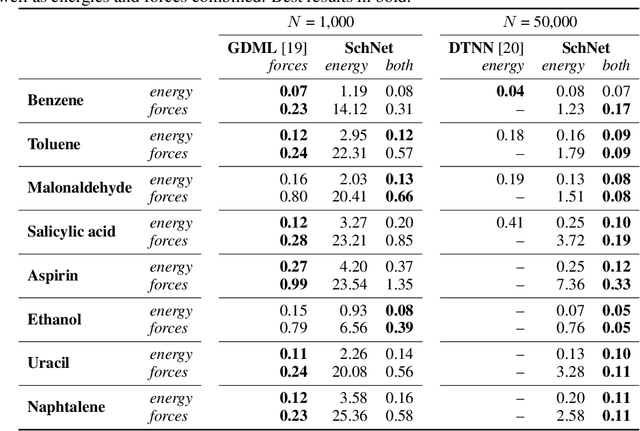
Deep learning has the potential to revolutionize quantum chemistry as it is ideally suited to learn representations for structured data and speed up the exploration of chemical space. While convolutional neural networks have proven to be the first choice for images, audio and video data, the atoms in molecules are not restricted to a grid. Instead, their precise locations contain essential physical information, that would get lost if discretized. Thus, we propose to use continuous-filter convolutional layers to be able to model local correlations without requiring the data to lie on a grid. We apply those layers in SchNet: a novel deep learning architecture modeling quantum interactions in molecules. We obtain a joint model for the total energy and interatomic forces that follows fundamental quantum-chemical principles. This includes rotationally invariant energy predictions and a smooth, differentiable potential energy surface. Our architecture achieves state-of-the-art performance for benchmarks of equilibrium molecules and molecular dynamics trajectories. Finally, we introduce a more challenging benchmark with chemical and structural variations that suggests the path for further work.