 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRelevant Walk Search for Explaining Graph Neural Networks
May 22, 2026Graph Neural Networks (GNNs) have become important machine learning tools for graph analysis, and its explainability is crucial for safety, fairness, and robustness. Layer-wise relevance propagation for GNNs (GNN-LRP) evaluates the relevance of \emph{walks} to reveal important information flows in the network, and provides higher-order explanations, which have been shown to be superior to the lower-order, i.e., node-/edge-level, explanations. However, identifying relevant walks by GNN-LRP requires {\em exponential} computational complexity with respect to the network depth, which we will remedy in this paper. Specifically, we propose {\em polynomial-time} algorithms for finding top-$K$ relevant walks, which drastically reduces the computation and thus increases the applicability of GNN-LRP to large-scale problems. Our proposed algorithms are based on the \emph{max-product} algorithm -- a common tool for finding the maximum likelihood configurations in probabilistic graphical models -- and can find the most relevant walks exactly at the neuron level and approximately at the node level. Our experiments demonstrate the performance of our algorithms at scale and their utility across application domains, i.e., on epidemiology, molecular, and natural language benchmarks. We provide our codes under \href{https://github.com/xiong-ping/rel_walk_gnnlrp}{github.com/xiong-ping/rel\_walk\_gnnlrp}.
* Published in ICML 2023
Enhanced Diffusion Sampling: Efficient Rare Event Sampling and Free Energy Calculation with Diffusion Models
Feb 18, 2026The rare-event sampling problem has long been the central limiting factor in molecular dynamics (MD), especially in biomolecular simulation. Recently, diffusion models such as BioEmu have emerged as powerful equilibrium samplers that generate independent samples from complex molecular distributions, eliminating the cost of sampling rare transition events. However, a sampling problem remains when computing observables that rely on states which are rare in equilibrium, for example folding free energies. Here, we introduce enhanced diffusion sampling, enabling efficient exploration of rare-event regions while preserving unbiased thermodynamic estimators. The key idea is to perform quantitatively accurate steering protocols to generate biased ensembles and subsequently recover equilibrium statistics via exact reweighting. We instantiate our framework in three algorithms: UmbrellaDiff (umbrella sampling with diffusion models), $Δ$G-Diff (free-energy differences via tilted ensembles), and MetaDiff (a batchwise analogue for metadynamics). Across toy systems, protein folding landscapes and folding free energies, our methods achieve fast, accurate, and scalable estimation of equilibrium properties within GPU-minutes to hours per system -- closing the rare-event sampling gap that remained after the advent of diffusion-model equilibrium samplers.
Improved motif-scaffolding with SE(3) flow matching
Jan 08, 2024Protein design often begins with knowledge of a desired function from a motif which motif-scaffolding aims to construct a functional protein around. Recently, generative models have achieved breakthrough success in designing scaffolds for a diverse range of motifs. However, the generated scaffolds tend to lack structural diversity, which can hinder success in wet-lab validation. In this work, we extend FrameFlow, an SE(3) flow matching model for protein backbone generation, to perform motif-scaffolding with two complementary approaches. The first is motif amortization, in which FrameFlow is trained with the motif as input using a data augmentation strategy. The second is motif guidance, which performs scaffolding using an estimate of the conditional score from FrameFlow, and requires no additional training. Both approaches achieve an equivalent or higher success rate than previous state-of-the-art methods, with 2.5 times more structurally diverse scaffolds. Code: https://github.com/ microsoft/frame-flow.
Scaling machine learning-based chemical plant simulation: A method for fine-tuning a model to induce stable fixed points
Jul 25, 2023Idealized first-principles models of chemical plants can be inaccurate. An alternative is to fit a Machine Learning (ML) model directly to plant sensor data. We use a structured approach: Each unit within the plant gets represented by one ML model. After fitting the models to the data, the models are connected into a flowsheet-like directed graph. We find that for smaller plants, this approach works well, but for larger plants, the complex dynamics arising from large and nested cycles in the flowsheet lead to instabilities in the cycle solver. We analyze this problem in depth and show that it is not merely a specialized concern but rather a more pervasive challenge that will likely occur whenever ML is applied to larger plants. To address this problem, we present a way to fine-tune ML models such that solving cycles with the usual methods becomes robust again.
SchNetPack 2.0: A neural network toolbox for atomistic machine learning
Dec 11, 2022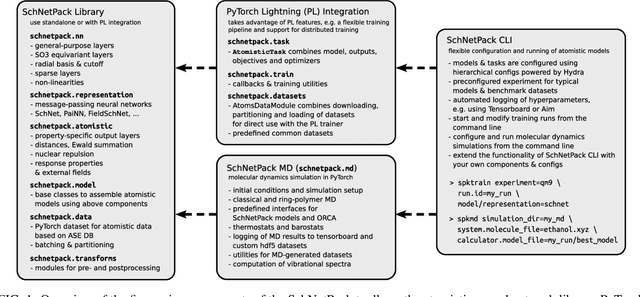
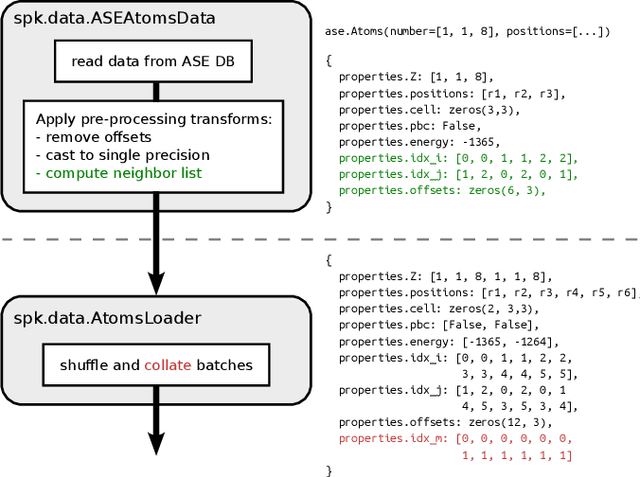
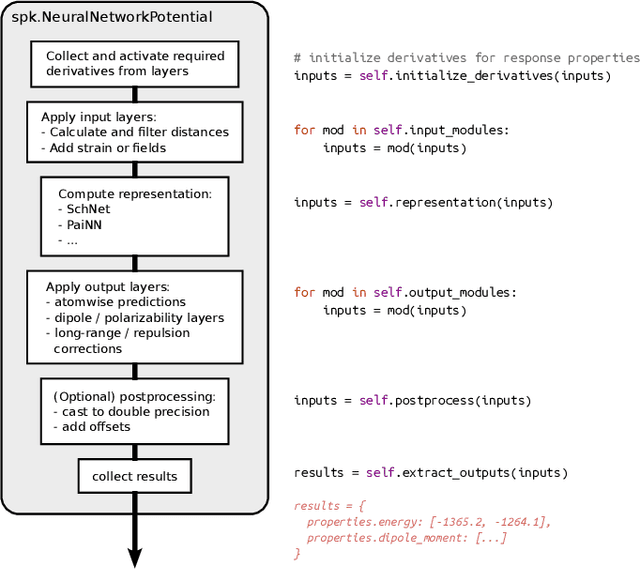

SchNetPack is a versatile neural networks toolbox that addresses both the requirements of method development and application of atomistic machine learning. Version 2.0 comes with an improved data pipeline, modules for equivariant neural networks as well as a PyTorch implementation of molecular dynamics. An optional integration with PyTorch Lightning and the Hydra configuration framework powers a flexible command-line interface. This makes SchNetPack 2.0 easily extendable with custom code and ready for complex training task such as generation of 3d molecular structures.
Accurate Machine Learned Quantum-Mechanical Force Fields for Biomolecular Simulations
May 17, 2022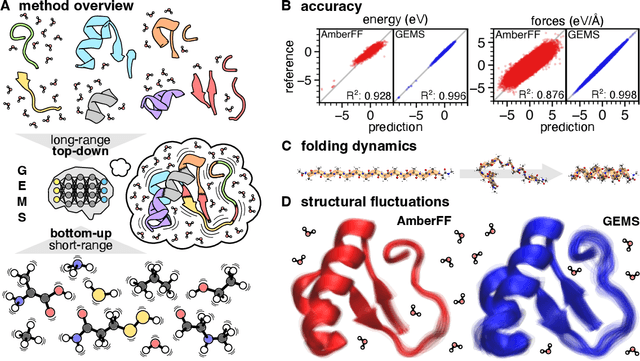
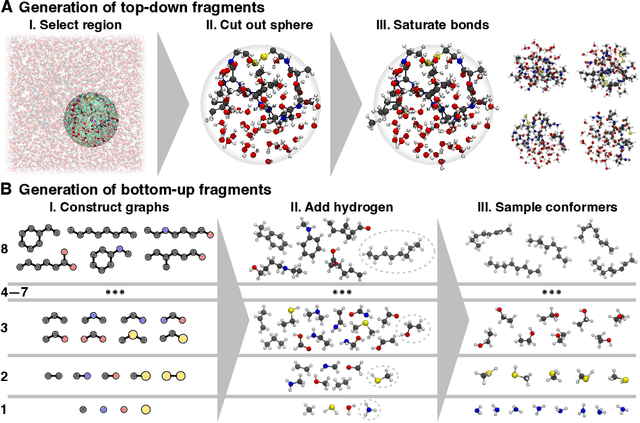
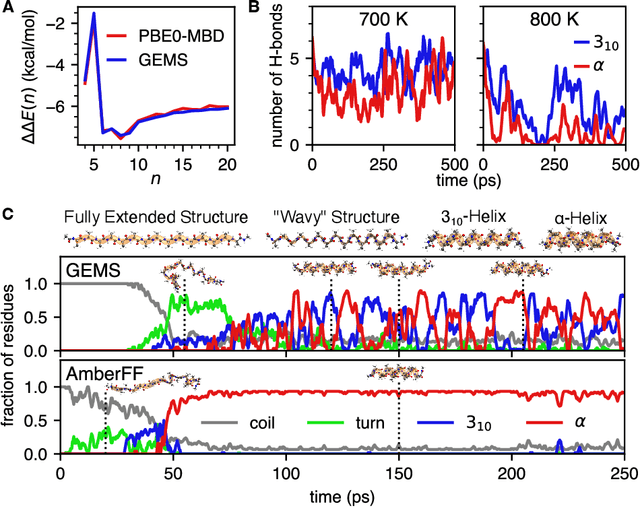
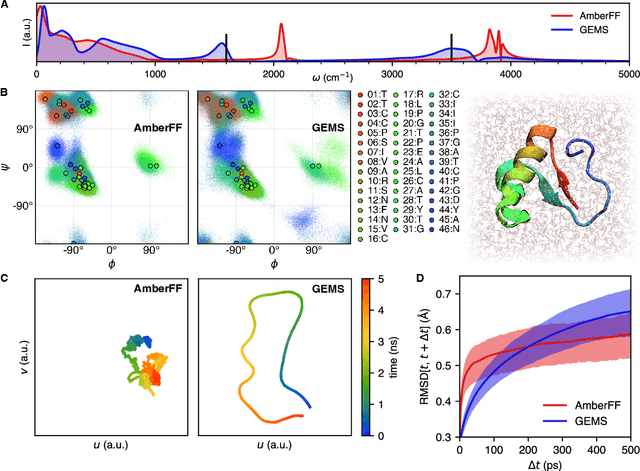
Molecular dynamics (MD) simulations allow atomistic insights into chemical and biological processes. Accurate MD simulations require computationally demanding quantum-mechanical calculations, being practically limited to short timescales and few atoms. For larger systems, efficient, but much less reliable empirical force fields are used. Recently, machine learned force fields (MLFFs) emerged as an alternative means to execute MD simulations, offering similar accuracy as ab initio methods at orders-of-magnitude speedup. Until now, MLFFs mainly capture short-range interactions in small molecules or periodic materials, due to the increased complexity of constructing models and obtaining reliable reference data for large molecules, where long-ranged many-body effects become important. This work proposes a general approach to constructing accurate MLFFs for large-scale molecular simulations (GEMS) by training on "bottom-up" and "top-down" molecular fragments of varying size, from which the relevant physicochemical interactions can be learned. GEMS is applied to study the dynamics of alanine-based peptides and the 46-residue protein crambin in aqueous solution, allowing nanosecond-scale MD simulations of >25k atoms at essentially ab initio quality. Our findings suggest that structural motifs in peptides and proteins are more flexible than previously thought, indicating that simulations at ab initio accuracy might be necessary to understand dynamic biomolecular processes such as protein (mis)folding, drug-protein binding, or allosteric regulation.
Automatic Identification of Chemical Moieties
Mar 30, 2022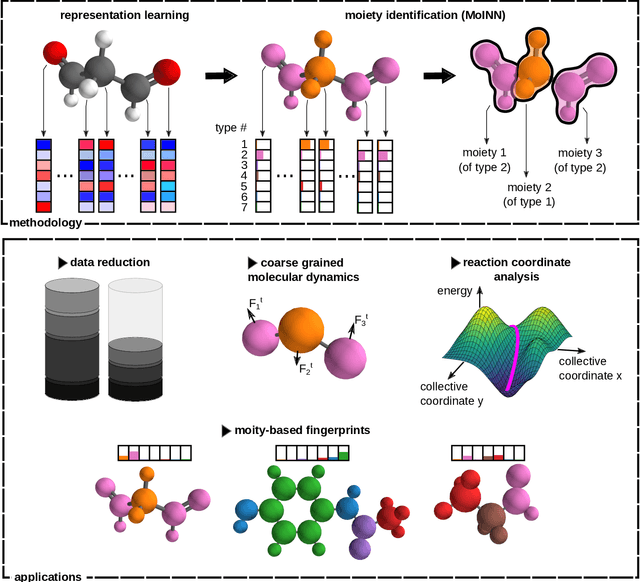
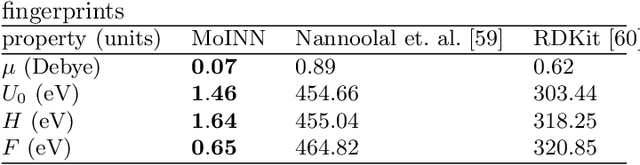
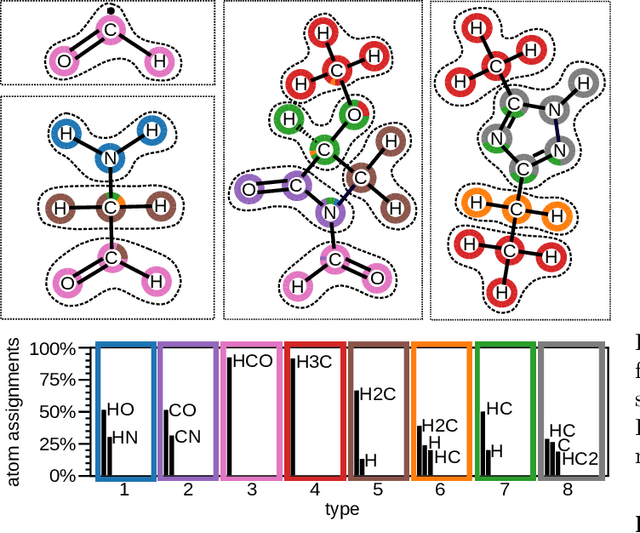
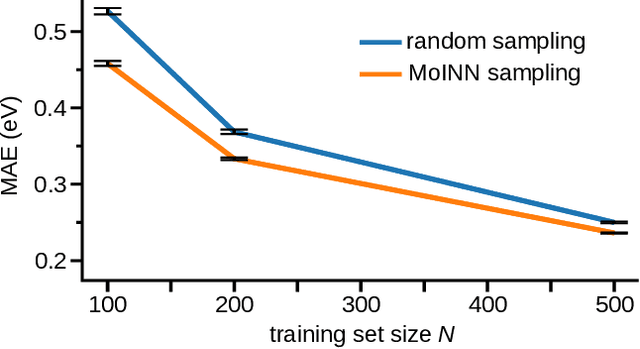
In recent years, the prediction of quantum mechanical observables with machine learning methods has become increasingly popular. Message-passing neural networks (MPNNs) solve this task by constructing atomic representations, from which the properties of interest are predicted. Here, we introduce a method to automatically identify chemical moieties (molecular building blocks) from such representations, enabling a variety of applications beyond property prediction, which otherwise rely on expert knowledge. The required representation can either be provided by a pretrained MPNN, or learned from scratch using only structural information. Beyond the data-driven design of molecular fingerprints, the versatility of our approach is demonstrated by enabling the selection of representative entries in chemical databases, the automatic construction of coarse-grained force fields, as well as the identification of reaction coordinates.
Inverse design of 3d molecular structures with conditional generative neural networks
Sep 10, 2021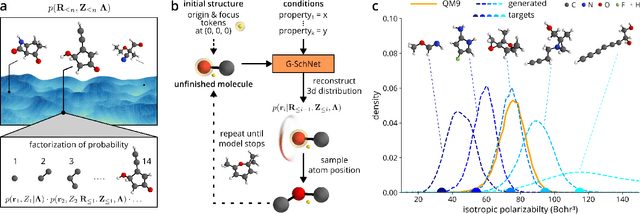
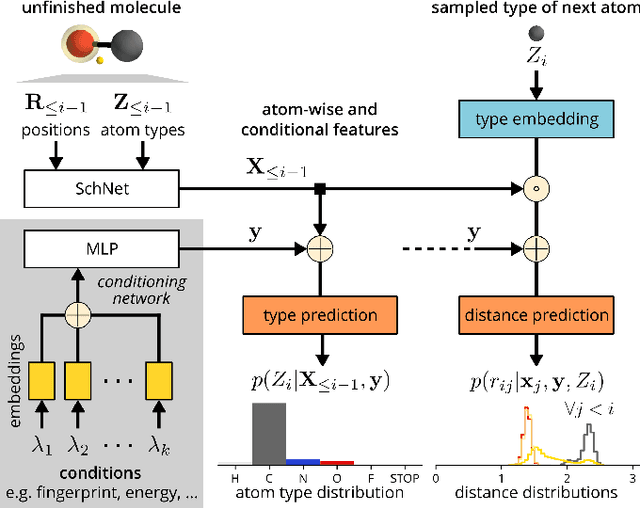
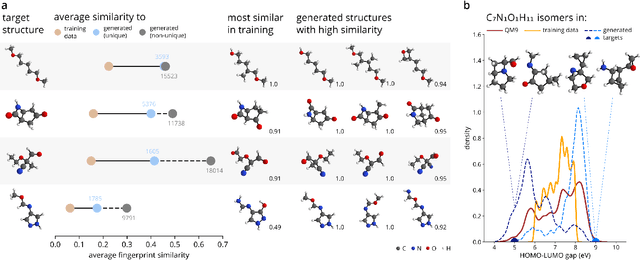
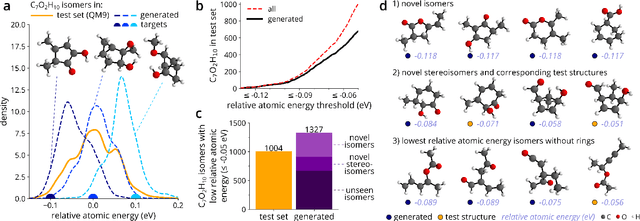
The rational design of molecules with desired properties is a long-standing challenge in chemistry. Generative neural networks have emerged as a powerful approach to sample novel molecules from a learned distribution. Here, we propose a conditional generative neural network for 3d molecular structures with specified structural and chemical properties. This approach is agnostic to chemical bonding and enables targeted sampling of novel molecules from conditional distributions, even in domains where reference calculations are sparse. We demonstrate the utility of our method for inverse design by generating molecules with specified composition or motifs, discovering particularly stable molecules, and jointly targeting multiple electronic properties beyond the training regime.
SE(3)-equivariant prediction of molecular wavefunctions and electronic densities
Jun 04, 2021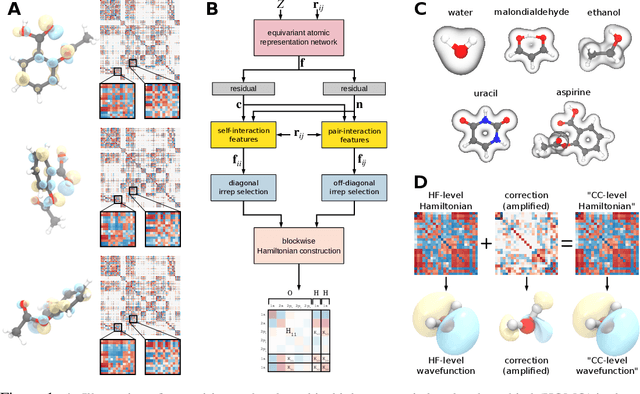
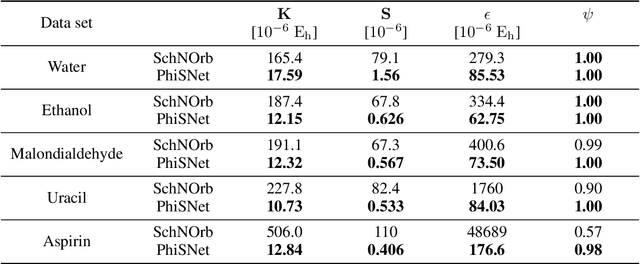
Machine learning has enabled the prediction of quantum chemical properties with high accuracy and efficiency, allowing to bypass computationally costly ab initio calculations. Instead of training on a fixed set of properties, more recent approaches attempt to learn the electronic wavefunction (or density) as a central quantity of atomistic systems, from which all other observables can be derived. This is complicated by the fact that wavefunctions transform non-trivially under molecular rotations, which makes them a challenging prediction target. To solve this issue, we introduce general SE(3)-equivariant operations and building blocks for constructing deep learning architectures for geometric point cloud data and apply them to reconstruct wavefunctions of atomistic systems with unprecedented accuracy. Our model reduces prediction errors by up to two orders of magnitude compared to the previous state-of-the-art and makes it possible to derive properties such as energies and forces directly from the wavefunction in an end-to-end manner. We demonstrate the potential of our approach in a transfer learning application, where a model trained on low accuracy reference wavefunctions implicitly learns to correct for electronic many-body interactions from observables computed at a higher level of theory. Such machine-learned wavefunction surrogates pave the way towards novel semi-empirical methods, offering resolution at an electronic level while drastically decreasing computational cost. While we focus on physics applications in this contribution, the proposed equivariant framework for deep learning on point clouds is promising also beyond, say, in computer vision or graphics.
SpookyNet: Learning Force Fields with Electronic Degrees of Freedom and Nonlocal Effects
May 01, 2021



In recent years, machine-learned force fields (ML-FFs) have gained increasing popularity in the field of computational chemistry. Provided they are trained on appropriate reference data, ML-FFs combine the accuracy of ab initio methods with the efficiency of conventional force fields. However, current ML-FFs typically ignore electronic degrees of freedom, such as the total charge or spin, when forming their prediction. In addition, they often assume chemical locality, which can be problematic in cases where nonlocal effects play a significant role. This work introduces SpookyNet, a deep neural network for constructing ML-FFs with explicit treatment of electronic degrees of freedom and quantum nonlocality. Its predictions are further augmented with physically-motivated corrections to improve the description of long-ranged interactions and nuclear repulsion. SpookyNet improves upon the current state-of-the-art (or achieves similar performance) on popular quantum chemistry data sets. Notably, it can leverage the learned chemical insights, e.g. by predicting unknown spin states or by properly modeling physical limits. Moreover, it is able to generalize across chemical and conformational space and thus close an important remaining gap for today's machine learning models in quantum chemistry.