 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSchNetPack 2.0: A neural network toolbox for atomistic machine learning
Dec 11, 2022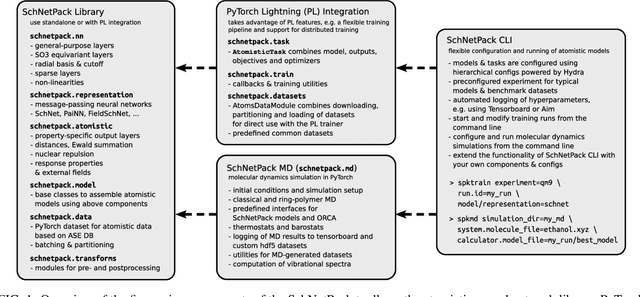
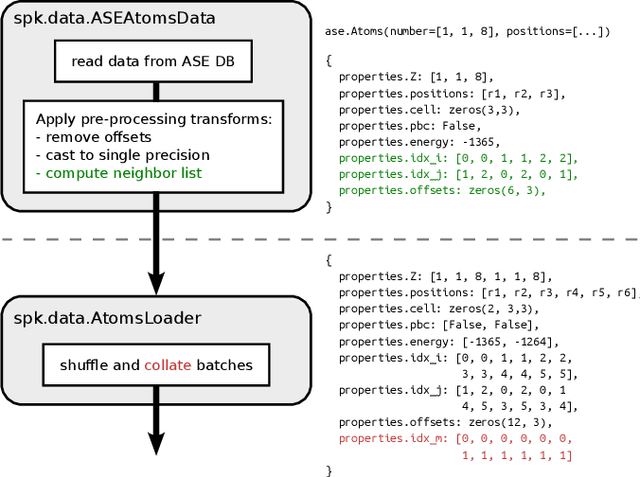
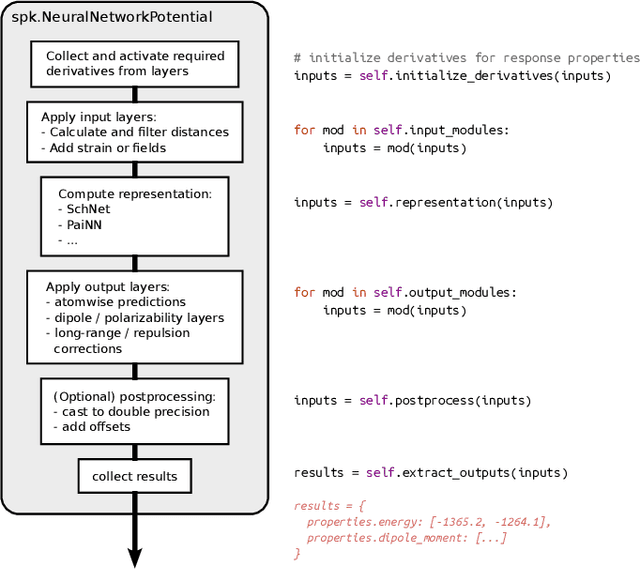

SchNetPack is a versatile neural networks toolbox that addresses both the requirements of method development and application of atomistic machine learning. Version 2.0 comes with an improved data pipeline, modules for equivariant neural networks as well as a PyTorch implementation of molecular dynamics. An optional integration with PyTorch Lightning and the Hydra configuration framework powers a flexible command-line interface. This makes SchNetPack 2.0 easily extendable with custom code and ready for complex training task such as generation of 3d molecular structures.
Inverse design of 3d molecular structures with conditional generative neural networks
Sep 10, 2021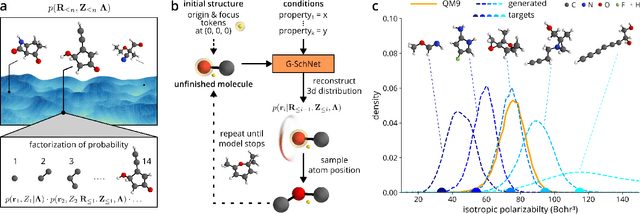
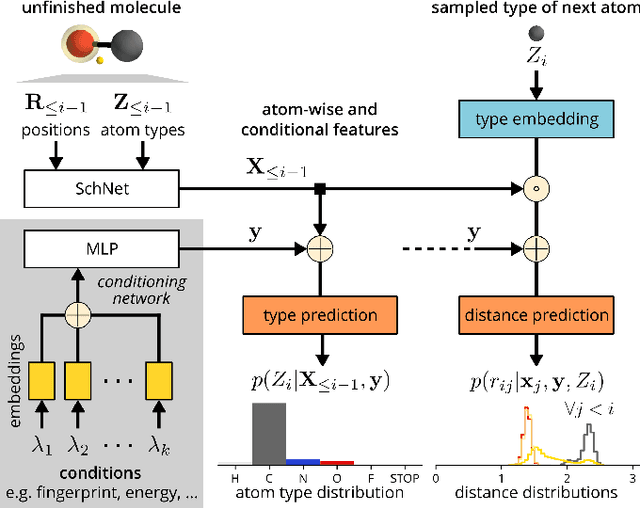
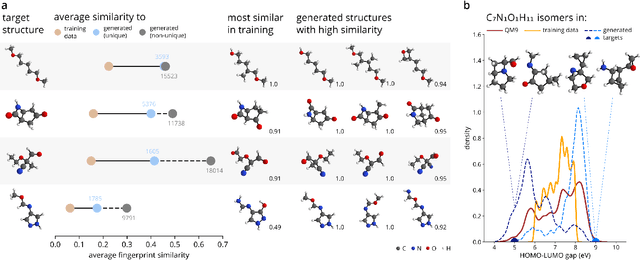
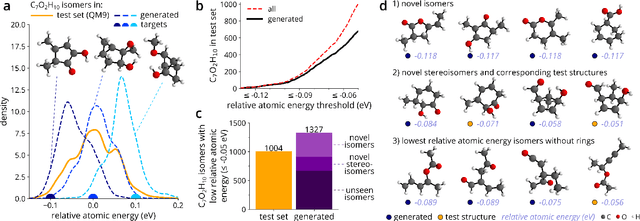
The rational design of molecules with desired properties is a long-standing challenge in chemistry. Generative neural networks have emerged as a powerful approach to sample novel molecules from a learned distribution. Here, we propose a conditional generative neural network for 3d molecular structures with specified structural and chemical properties. This approach is agnostic to chemical bonding and enables targeted sampling of novel molecules from conditional distributions, even in domains where reference calculations are sparse. We demonstrate the utility of our method for inverse design by generating molecules with specified composition or motifs, discovering particularly stable molecules, and jointly targeting multiple electronic properties beyond the training regime.
Symmetry-adapted generation of 3d point sets for the targeted discovery of molecules
Jun 02, 2019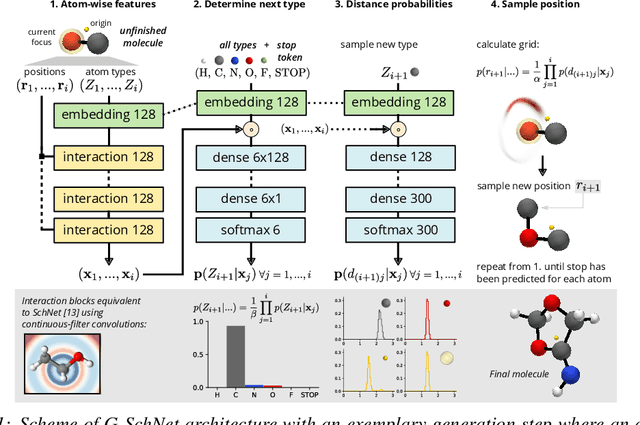
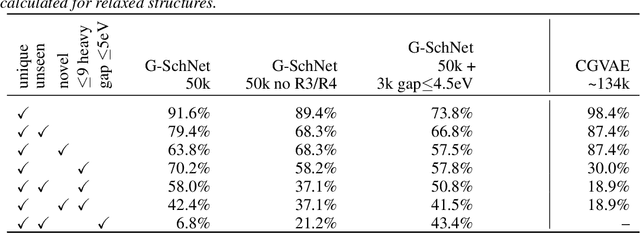
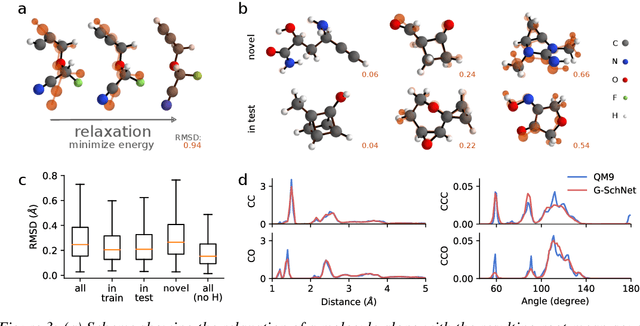
Deep learning has proven to yield fast and accurate predictions of quantum-chemical properties to accelerate the discovery of novel molecules and materials. As an exhaustive exploration of the vast chemical space is still infeasible, we require generative models that guide our search towards systems with desired properties. While graph-based models have previously been proposed, they are restricted by a lack of spatial information such that they are unable to recognize spatial isomerism and non-bonded interactions. Here, we introduce a generative neural network for 3d point sets that respects the rotational invariance of the targeted structures. We apply it to the generation of molecules and demonstrate its ability to approximate the distribution of equilibrium structures using spatial metrics as well as established measures from chemoinformatics. As our model is able to capture the complex relationship between 3d geometry and electronic properties, we bias the distribution of the generator towards molecules with a small HOMO-LUMO gap - an important property for the design of organic solar cells.
Generating equilibrium molecules with deep neural networks
Oct 26, 2018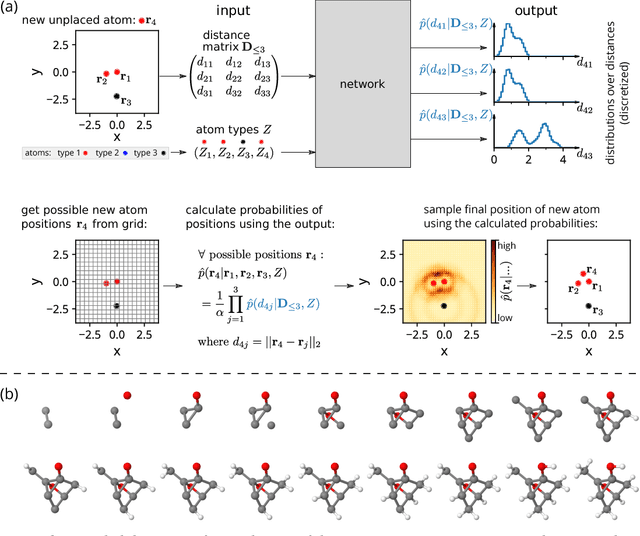
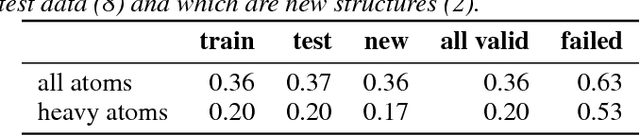
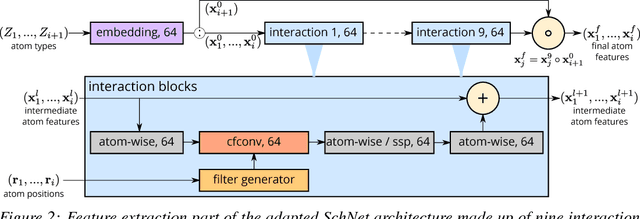
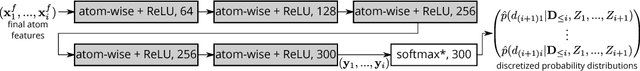
Discovery of atomistic systems with desirable properties is a major challenge in chemistry and material science. Here we introduce a novel, autoregressive, convolutional deep neural network architecture that generates molecular equilibrium structures by sequentially placing atoms in three-dimensional space. The model estimates the joint probability over molecular configurations with tractable conditional probabilities which only depend on distances between atoms and their nuclear charges. It combines concepts from state-of-the-art atomistic neural networks with auto-regressive generative models for images and speech. We demonstrate that the architecture is capable of generating molecules close to equilibrium for constitutional isomers of C$_7$O$_2$H$_{10}$.