 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeComplete and Efficient Covariants for 3D Point Configurations with Application to Learning Molecular Quantum Properties
Sep 04, 2024
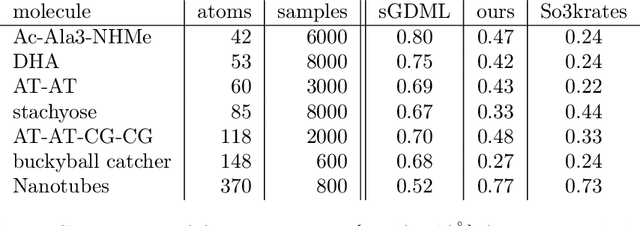
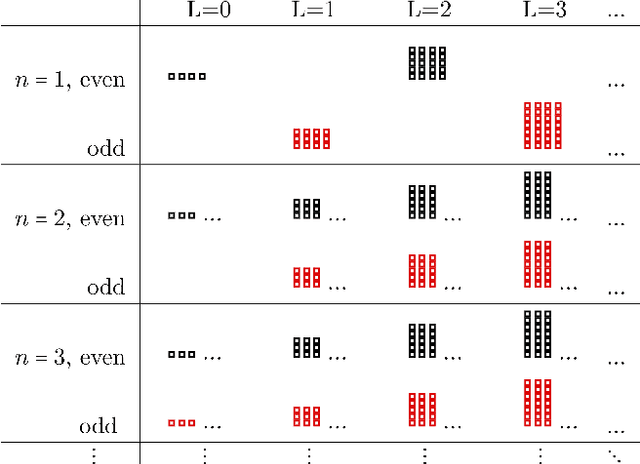

When modeling physical properties of molecules with machine learning, it is desirable to incorporate $SO(3)$-covariance. While such models based on low body order features are not complete, we formulate and prove general completeness properties for higher order methods, and show that $6k-5$ of these features are enough for up to $k$ atoms. We also find that the Clebsch--Gordan operations commonly used in these methods can be replaced by matrix multiplications without sacrificing completeness, lowering the scaling from $O(l^6)$ to $O(l^3)$ in the degree of the features. We apply this to quantum chemistry, but the proposed methods are generally applicable for problems involving 3D point configurations.
E3x: $\mathrm{E}$-Equivariant Deep Learning Made Easy
Jan 17, 2024
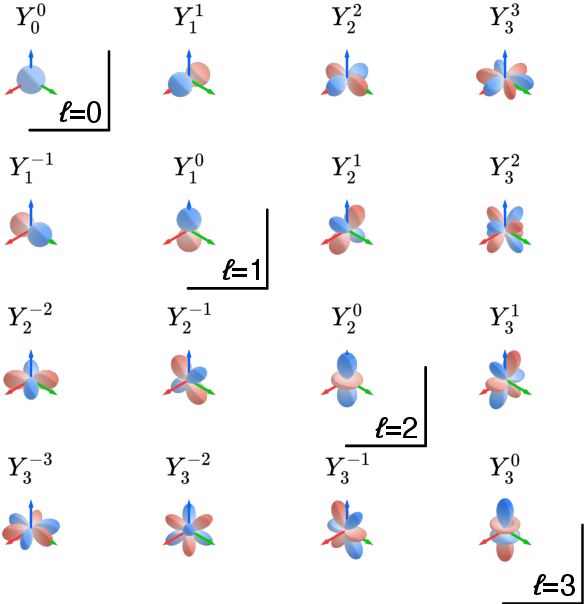
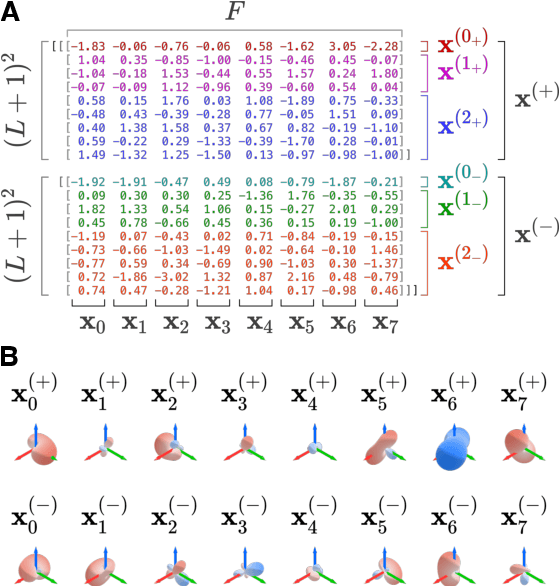

This work introduces E3x, a software package for building neural networks that are equivariant with respect to the Euclidean group $\mathrm{E}(3)$, consisting of translations, rotations, and reflections of three-dimensional space. Compared to ordinary neural networks, $\mathrm{E}(3)$-equivariant models promise benefits whenever input and/or output data are quantities associated with three-dimensional objects. This is because the numeric values of such quantities (e.g. positions) typically depend on the chosen coordinate system. Under transformations of the reference frame, the values change predictably, but the underlying rules can be difficult to learn for ordinary machine learning models. With built-in $\mathrm{E}(3)$-equivariance, neural networks are guaranteed to satisfy the relevant transformation rules exactly, resulting in superior data efficiency and accuracy. The code for E3x is available from https://github.com/google-research/e3x, detailed documentation and usage examples can be found on https://e3x.readthedocs.io.
Accurate Machine Learned Quantum-Mechanical Force Fields for Biomolecular Simulations
May 17, 2022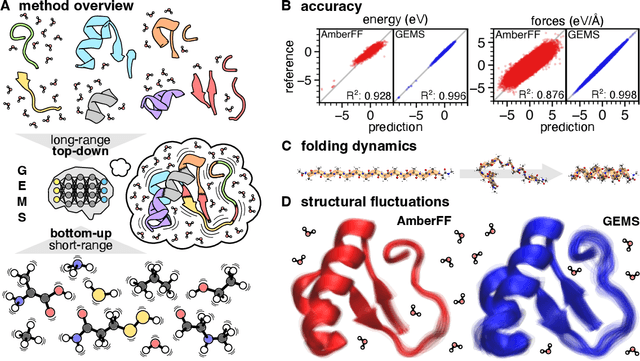
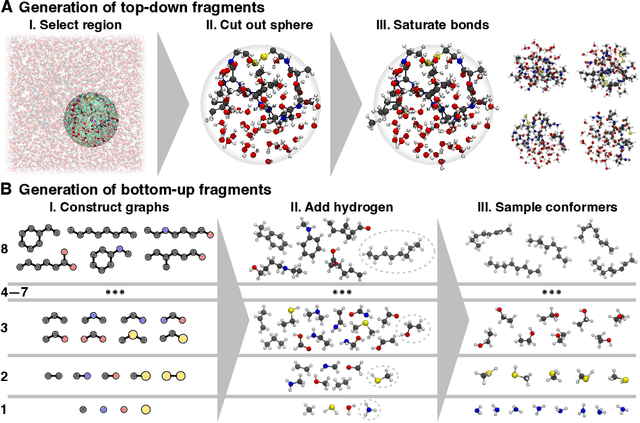
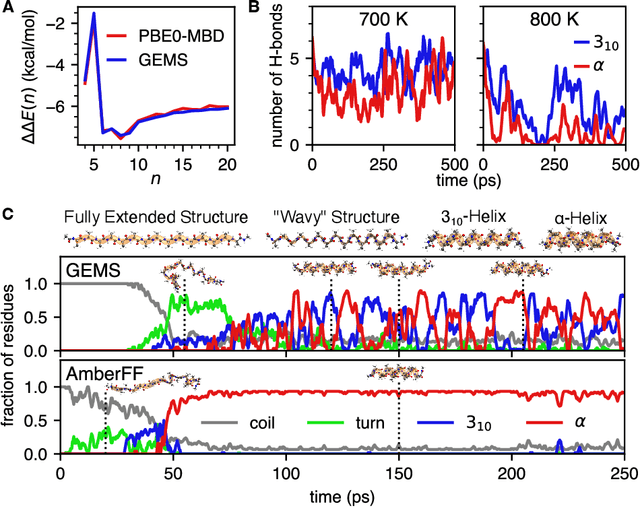
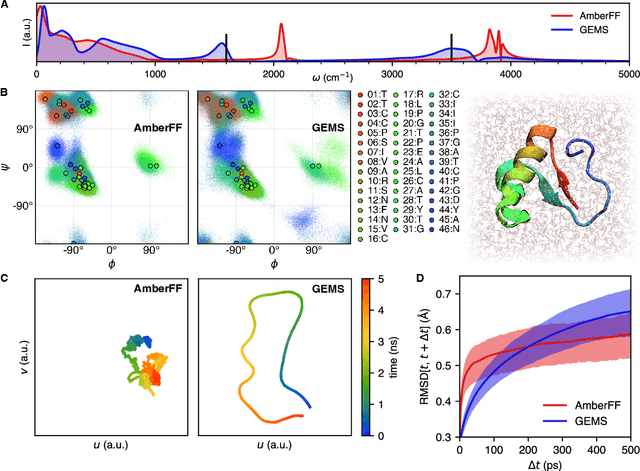
Molecular dynamics (MD) simulations allow atomistic insights into chemical and biological processes. Accurate MD simulations require computationally demanding quantum-mechanical calculations, being practically limited to short timescales and few atoms. For larger systems, efficient, but much less reliable empirical force fields are used. Recently, machine learned force fields (MLFFs) emerged as an alternative means to execute MD simulations, offering similar accuracy as ab initio methods at orders-of-magnitude speedup. Until now, MLFFs mainly capture short-range interactions in small molecules or periodic materials, due to the increased complexity of constructing models and obtaining reliable reference data for large molecules, where long-ranged many-body effects become important. This work proposes a general approach to constructing accurate MLFFs for large-scale molecular simulations (GEMS) by training on "bottom-up" and "top-down" molecular fragments of varying size, from which the relevant physicochemical interactions can be learned. GEMS is applied to study the dynamics of alanine-based peptides and the 46-residue protein crambin in aqueous solution, allowing nanosecond-scale MD simulations of >25k atoms at essentially ab initio quality. Our findings suggest that structural motifs in peptides and proteins are more flexible than previously thought, indicating that simulations at ab initio accuracy might be necessary to understand dynamic biomolecular processes such as protein (mis)folding, drug-protein binding, or allosteric regulation.
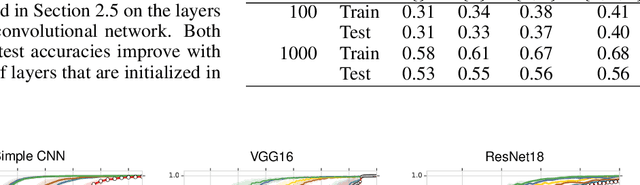
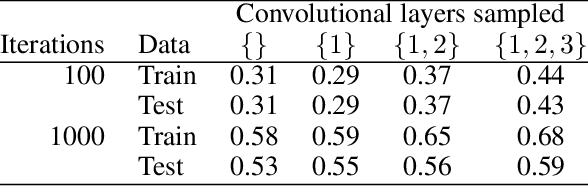
The Impact of Reinitialization on Generalization in Convolutional Neural Networks
Sep 01, 2021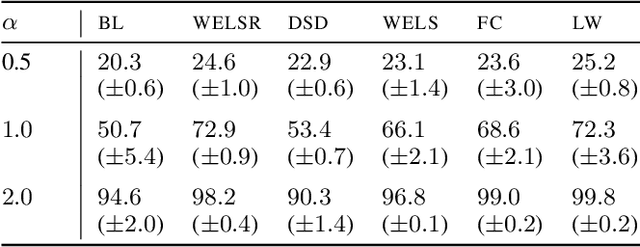



Recent results suggest that reinitializing a subset of the parameters of a neural network during training can improve generalization, particularly for small training sets. We study the impact of different reinitialization methods in several convolutional architectures across 12 benchmark image classification datasets, analyzing their potential gains and highlighting limitations. We also introduce a new layerwise reinitialization algorithm that outperforms previous methods and suggest explanations of the observed improved generalization. First, we show that layerwise reinitialization increases the margin on the training examples without increasing the norm of the weights, hence leading to an improvement in margin-based generalization bounds for neural networks. Second, we demonstrate that it settles in flatter local minima of the loss surface. Third, it encourages learning general rules and discourages memorization by placing emphasis on the lower layers of the neural network. Our takeaway message is that the accuracy of convolutional neural networks can be improved for small datasets using bottom-up layerwise reinitialization, where the number of reinitialized layers may vary depending on the available compute budget.
Deep Learning Through the Lens of Example Difficulty
Jun 18, 2021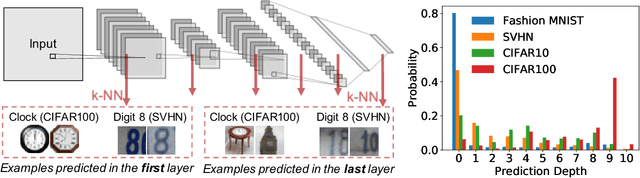


Existing work on understanding deep learning often employs measures that compress all data-dependent information into a few numbers. In this work, we adopt a perspective based on the role of individual examples. We introduce a measure of the computational difficulty of making a prediction for a given input: the (effective) prediction depth. Our extensive investigation reveals surprising yet simple relationships between the prediction depth of a given input and the model's uncertainty, confidence, accuracy and speed of learning for that data point. We further categorize difficult examples into three interpretable groups, demonstrate how these groups are processed differently inside deep models and showcase how this understanding allows us to improve prediction accuracy. Insights from our study lead to a coherent view of a number of separately reported phenomena in the literature: early layers generalize while later layers memorize; early layers converge faster and networks learn easy data and simple functions first.
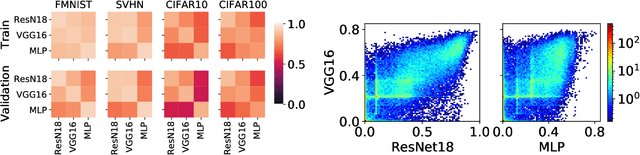
What Do Neural Networks Learn When Trained With Random Labels?
Jun 18, 2020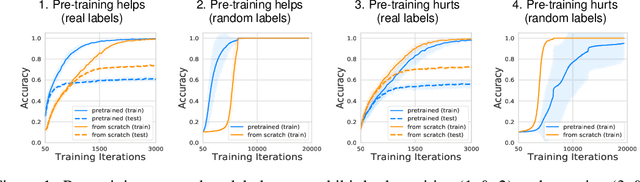
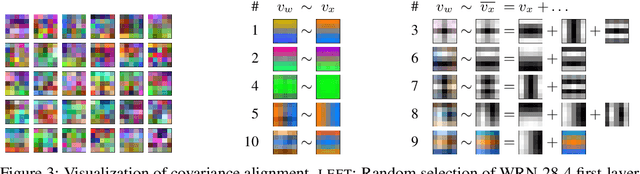
We study deep neural networks (DNNs) trained on natural image data with entirely random labels. Despite its popularity in the literature, where it is often used to study memorization, generalization, and other phenomena, little is known about what DNNs learn in this setting. In this paper, we show analytically for convolutional and fully connected networks that an alignment between the principal components of network parameters and data takes place when training with random labels. We study this alignment effect by investigating neural networks pre-trained on randomly labelled image data and subsequently fine-tuned on disjoint datasets with random or real labels. We show how this alignment produces a positive transfer: networks pre-trained with random labels train faster downstream compared to training from scratch even after accounting for simple effects, such as weight scaling. We analyze how competing effects, such as specialization at later layers, may hide the positive transfer. These effects are studied in several network architectures, including VGG16 and ResNet18, on CIFAR10 and ImageNet.
Exact marginal inference in Latent Dirichlet Allocation
Mar 31, 2020Assume we have potential "causes" $z\in Z$, which produce "events" $w$ with known probabilities $\beta(w|z)$. We observe $w_1,w_2,...,w_n$, what can we say about the distribution of the causes? A Bayesian estimate will assume a prior on distributions on $Z$ (we assume a Dirichlet prior) and calculate a posterior. An average over that posterior then gives a distribution on $Z$, which estimates how much each cause $z$ contributed to our observations. This is the setting of Latent Dirichlet Allocation, which can be applied e.g. to topics "producing" words in a document. In this setting usually the number of observed words is large, but the number of potential topics is small. We are here interested in applications with many potential "causes" (e.g. locations on the globe), but only a few observations. We show that the exact Bayesian estimate can be computed in linear time (and constant space) in $|Z|$ for a given upper bound on $n$ with a surprisingly simple formula. We generalize this algorithm to the case of sparse probabilities $\beta(w|z)$, in which we only need to assume that the tree width of an "interaction graph" on the observations is limited. On the other hand we also show that without such limitation the problem is NP-hard.
Adaptive Temporal-Difference Learning for Policy Evaluation with Per-State Uncertainty Estimates
Jun 19, 2019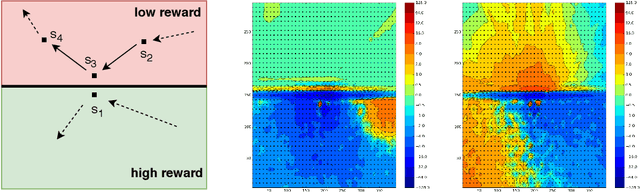

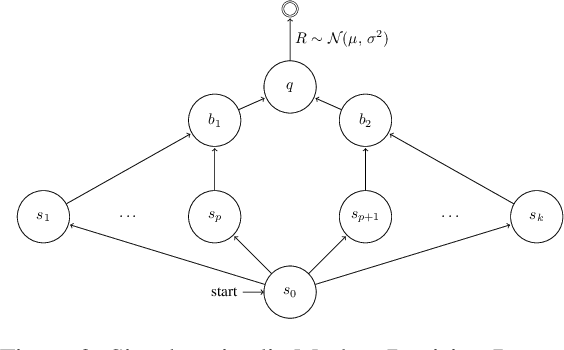
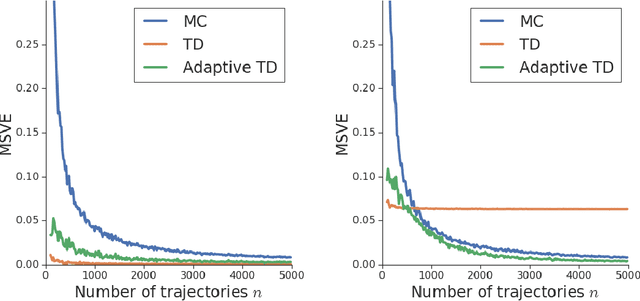
We consider the core reinforcement-learning problem of on-policy value function approximation from a batch of trajectory data, and focus on various issues of Temporal Difference (TD) learning and Monte Carlo (MC) policy evaluation. The two methods are known to achieve complementary bias-variance trade-off properties, with TD tending to achieve lower variance but potentially higher bias. In this paper, we argue that the larger bias of TD can be a result of the amplification of local approximation errors. We address this by proposing an algorithm that adaptively switches between TD and MC in each state, thus mitigating the propagation of errors. Our method is based on learned confidence intervals that detect biases of TD estimates. We demonstrate in a variety of policy evaluation tasks that this simple adaptive algorithm performs competitively with the best approach in hindsight, suggesting that learned confidence intervals are a powerful technique for adapting policy evaluation to use TD or MC returns in a data-driven way.
Temporal Difference Learning with Neural Networks - Study of the Leakage Propagation Problem
Jul 09, 2018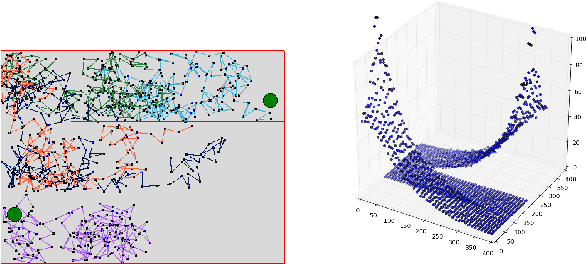
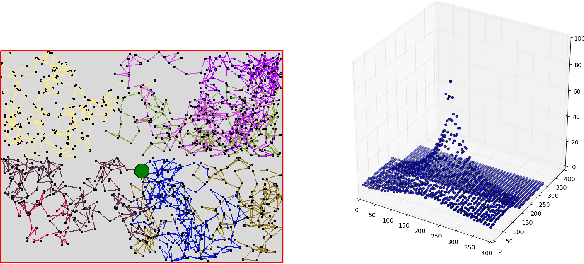


Temporal-Difference learning (TD) [Sutton, 1988] with function approximation can converge to solutions that are worse than those obtained by Monte-Carlo regression, even in the simple case of on-policy evaluation. To increase our understanding of the problem, we investigate the issue of approximation errors in areas of sharp discontinuities of the value function being further propagated by bootstrap updates. We show empirical evidence of this leakage propagation, and show analytically that it must occur, in a simple Markov chain, when function approximation errors are present. For reversible policies, the result can be interpreted as the tension between two terms of the loss function that TD minimises, as recently described by [Ollivier, 2018]. We show that the upper bounds from [Tsitsiklis and Van Roy, 1997] hold, but they do not imply that leakage propagation occurs and under what conditions. Finally, we test whether the problem could be mitigated with a better state representation, and whether it can be learned in an unsupervised manner, without rewards or privileged information.
Gradient Descent Quantizes ReLU Network Features
Mar 22, 2018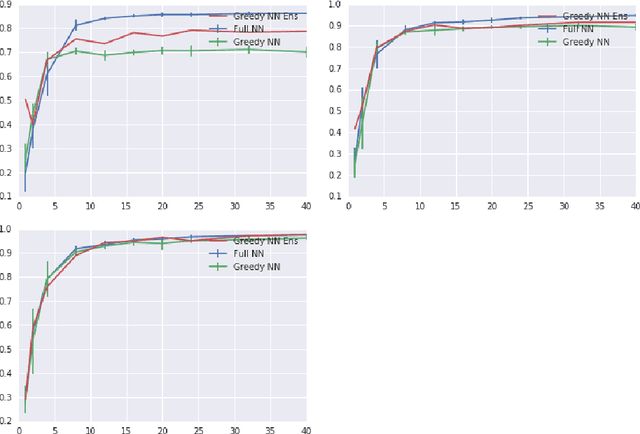
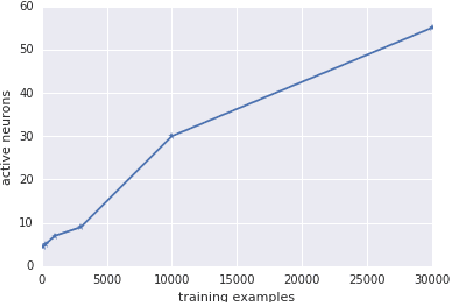
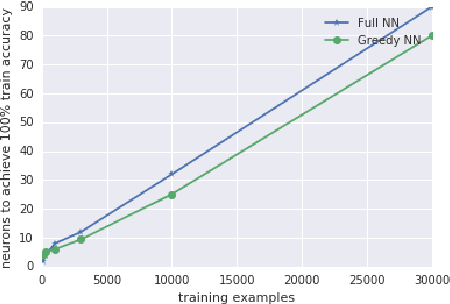
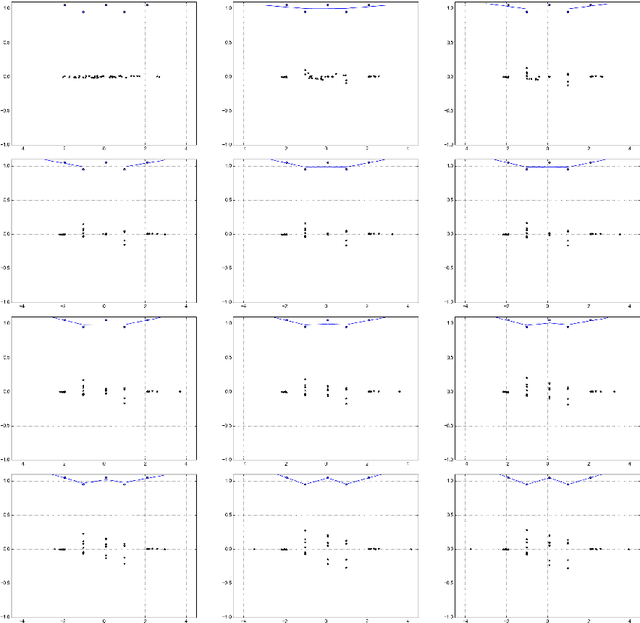
Deep neural networks are often trained in the over-parametrized regime (i.e. with far more parameters than training examples), and understanding why the training converges to solutions that generalize remains an open problem. Several studies have highlighted the fact that the training procedure, i.e. mini-batch Stochastic Gradient Descent (SGD) leads to solutions that have specific properties in the loss landscape. However, even with plain Gradient Descent (GD) the solutions found in the over-parametrized regime are pretty good and this phenomenon is poorly understood. We propose an analysis of this behavior for feedforward networks with a ReLU activation function under the assumption of small initialization and learning rate and uncover a quantization effect: The weight vectors tend to concentrate at a small number of directions determined by the input data. As a consequence, we show that for given input data there are only finitely many, "simple" functions that can be obtained, independent of the network size. This puts these functions in analogy to linear interpolations (for given input data there are finitely many triangulations, which each determine a function by linear interpolation). We ask whether this analogy extends to the generalization properties - while the usual distribution-independent generalization property does not hold, it could be that for e.g. smooth functions with bounded second derivative an approximation property holds which could "explain" generalization of networks (of unbounded size) to unseen inputs.