 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHow simple can you go? An off-the-shelf transformer approach to molecular dynamics
Mar 05, 2025Most current neural networks for molecular dynamics (MD) include physical inductive biases, resulting in specialized and complex architectures. This is in contrast to most other machine learning domains, where specialist approaches are increasingly replaced by general-purpose architectures trained on vast datasets. In line with this trend, several recent studies have questioned the necessity of architectural features commonly found in MD models, such as built-in rotational equivariance or energy conservation. In this work, we contribute to the ongoing discussion by evaluating the performance of an MD model with as few specialized architectural features as possible. We present a recipe for MD using an Edge Transformer, an "off-the-shelf'' transformer architecture that has been minimally modified for the MD domain, termed MD-ET. Our model implements neither built-in equivariance nor energy conservation. We use a simple supervised pre-training scheme on $\sim$30 million molecular structures from the QCML database. Using this "off-the-shelf'' approach, we show state-of-the-art results on several benchmarks after fine-tuning for a small number of steps. Additionally, we examine the effects of being only approximately equivariant and energy conserving for MD simulations, proposing a novel method for distinguishing the errors resulting from non-equivariance from other sources of inaccuracies like numerical rounding errors. While our model exhibits runaway energy increases on larger structures, we show approximately energy-conserving NVE simulations for a range of small structures.
Accurate Machine Learned Quantum-Mechanical Force Fields for Biomolecular Simulations
May 17, 2022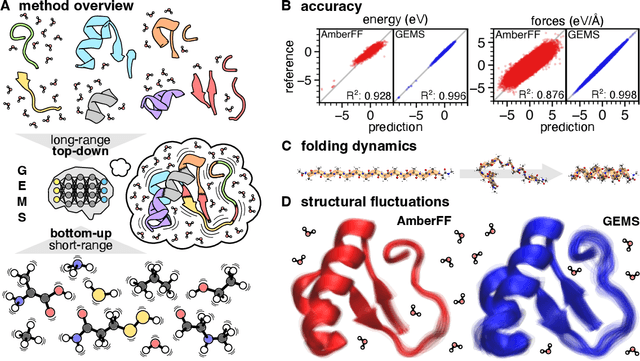
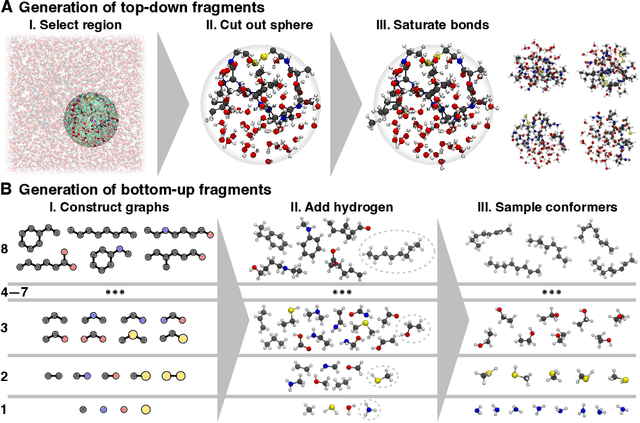
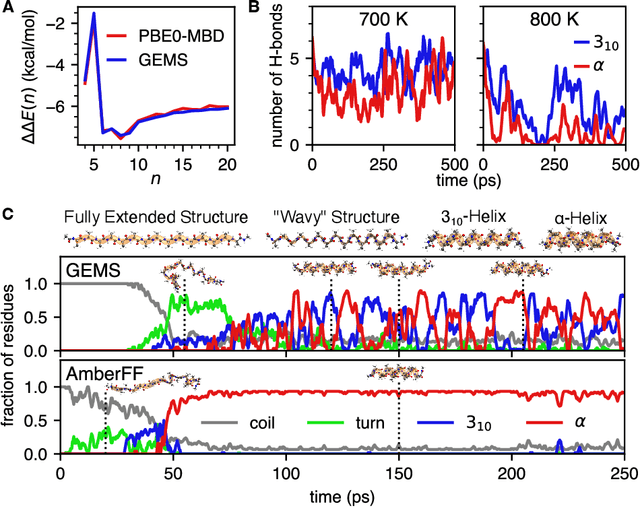
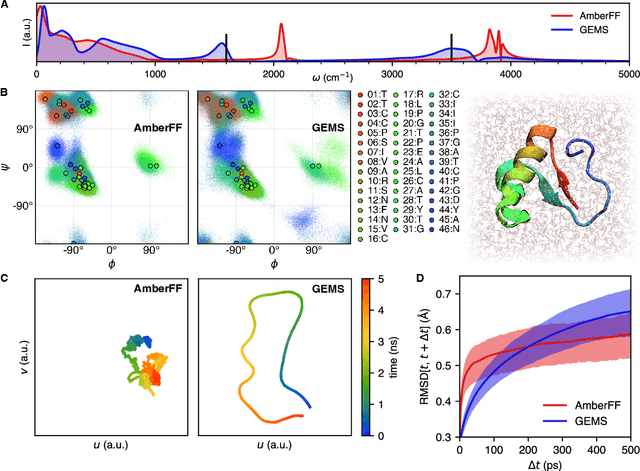
Molecular dynamics (MD) simulations allow atomistic insights into chemical and biological processes. Accurate MD simulations require computationally demanding quantum-mechanical calculations, being practically limited to short timescales and few atoms. For larger systems, efficient, but much less reliable empirical force fields are used. Recently, machine learned force fields (MLFFs) emerged as an alternative means to execute MD simulations, offering similar accuracy as ab initio methods at orders-of-magnitude speedup. Until now, MLFFs mainly capture short-range interactions in small molecules or periodic materials, due to the increased complexity of constructing models and obtaining reliable reference data for large molecules, where long-ranged many-body effects become important. This work proposes a general approach to constructing accurate MLFFs for large-scale molecular simulations (GEMS) by training on "bottom-up" and "top-down" molecular fragments of varying size, from which the relevant physicochemical interactions can be learned. GEMS is applied to study the dynamics of alanine-based peptides and the 46-residue protein crambin in aqueous solution, allowing nanosecond-scale MD simulations of >25k atoms at essentially ab initio quality. Our findings suggest that structural motifs in peptides and proteins are more flexible than previously thought, indicating that simulations at ab initio accuracy might be necessary to understand dynamic biomolecular processes such as protein (mis)folding, drug-protein binding, or allosteric regulation.