 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScaling machine learning-based chemical plant simulation: A method for fine-tuning a model to induce stable fixed points
Jul 25, 2023Idealized first-principles models of chemical plants can be inaccurate. An alternative is to fit a Machine Learning (ML) model directly to plant sensor data. We use a structured approach: Each unit within the plant gets represented by one ML model. After fitting the models to the data, the models are connected into a flowsheet-like directed graph. We find that for smaller plants, this approach works well, but for larger plants, the complex dynamics arising from large and nested cycles in the flowsheet lead to instabilities in the cycle solver. We analyze this problem in depth and show that it is not merely a specialized concern but rather a more pervasive challenge that will likely occur whenever ML is applied to larger plants. To address this problem, we present a way to fine-tune ML models such that solving cycles with the usual methods becomes robust again.
Autonomous robotic nanofabrication with reinforcement learning
Feb 27, 2020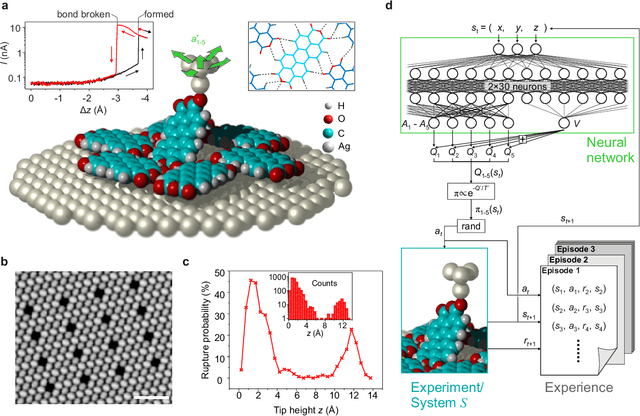
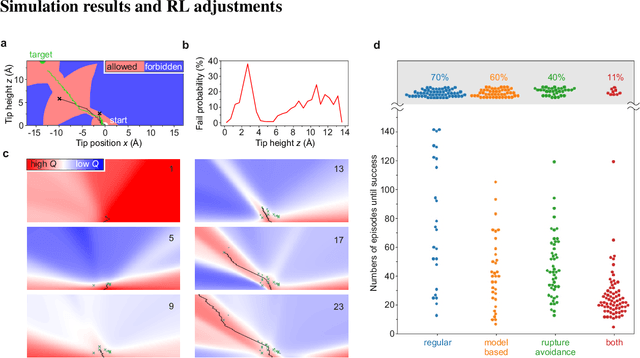
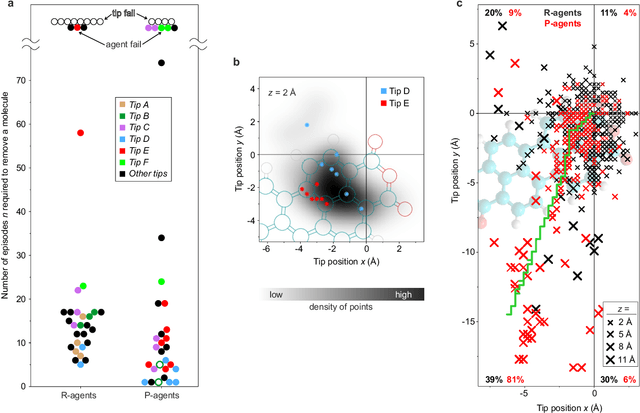
The ability to handle single molecules as effectively as macroscopic building-blocks would enable the construction of complex supramolecular structures that are not accessible by self-assembly. The fundamental challenges on the way towards this goal are the uncontrolled variability and poor observability of atomic-scale conformations. Here, we present a strategy to work around both obstacles, and demonstrate autonomous robotic nanofabrication by manipulating single molecules. Our approach employs reinforcement learning (RL), which is able to learn solution strategies even in the face of large uncertainty and with sparse feedback. However, to be useful for autonomous nanofabrication, standard RL algorithms need to be adapted to cope with the limited training opportunities available. We demonstrate the potential of our RL approach by applying it to an exemplary task of subtractive manufacturing, the removal of individual molecules from a molecular layer using a scanning probe microscope (SPM). Our RL agent reaches an excellent performance level, enabling us to automate a task which previously had to be performed by a human. We anticipate that our work opens the way towards autonomous agents for the robotic construction of functional supramolecular structures with speed, precision and perseverance beyond our current capabilities.
From Clustering to Cluster Explanations via Neural Networks
Jun 18, 2019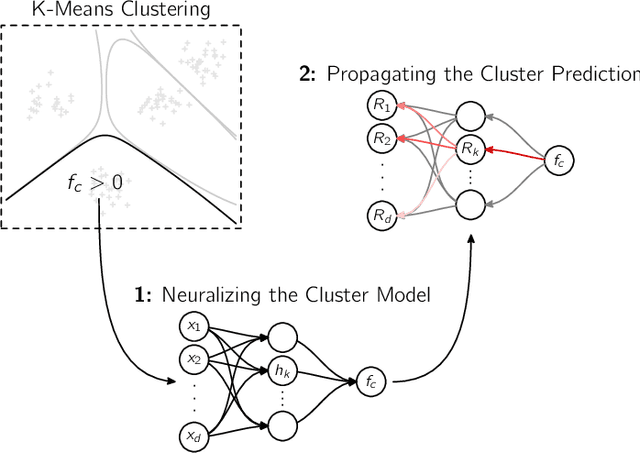
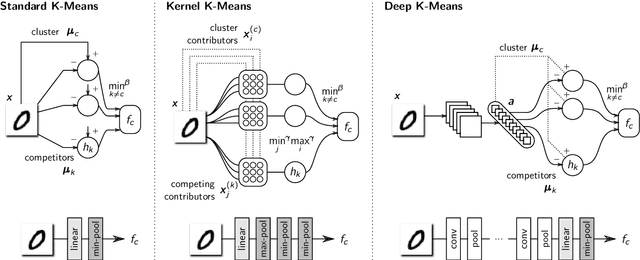
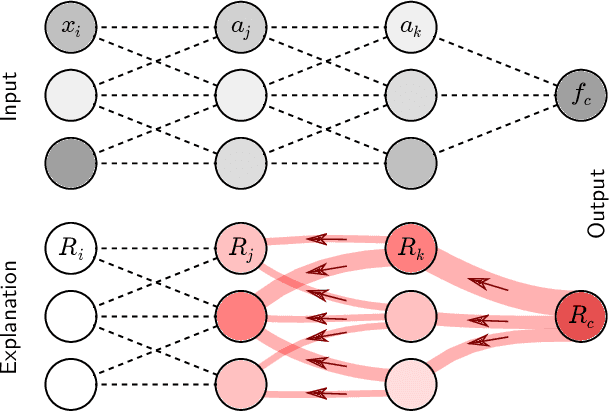

A wealth of algorithms have been developed to extract natural cluster structure in data. Identifying this structure is desirable but not always sufficient: We may also want to understand why the data points have been assigned to a given cluster. Clustering algorithms do not offer a systematic answer to this simple question. Hence we propose a new framework that can, for the first time, explain cluster assignments in terms of input features in a comprehensive manner. It is based on the novel theoretical insight that clustering models can be rewritten as neural networks, or 'neuralized'. Predictions of the obtained networks can then be quickly and accurately attributed to the input features. Several showcases demonstrate the ability of our method to assess the quality of learned clusters and to extract novel insights from the analyzed data and representations.