 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDe novo molecular structure elucidation from mass spectra via flow matching
Feb 23, 2026Mass spectrometry is a powerful and widely used tool for identifying molecular structures due to its sensitivity and ability to profile complex samples. However, translating spectra into full molecular structures is a difficult, under-defined inverse problem. Overcoming this problem is crucial for enabling biological insight, discovering new metabolites, and advancing chemical research across multiple fields. To this end, we develop MSFlow, a two-stage encoder-decoder flow-matching generative model that achieves state-of-the-art performance on the structure elucidation task for small molecules. In the first stage, we adopt a formula-restricted transformer model for encoding mass spectra into a continuous and chemically informative embedding space, while in the second stage, we train a decoder flow matching model to reconstruct molecules from latent embeddings of mass spectra. We present ablation studies demonstrating the importance of using information-preserving molecular descriptors for encoding mass spectra and motivate the use of our discrete flow-based decoder. Our rigorous evaluation demonstrates that MSFlow can accurately translate up to 45 percent of molecular mass spectra into their corresponding molecular representations - an improvement of up to fourteen-fold over the current state-of-the-art. A trained version of MSFlow is made publicly available on GitHub for non-commercial users.
FLOWR: Flow Matching for Structure-Aware De Novo, Interaction- and Fragment-Based Ligand Generation
Apr 14, 2025
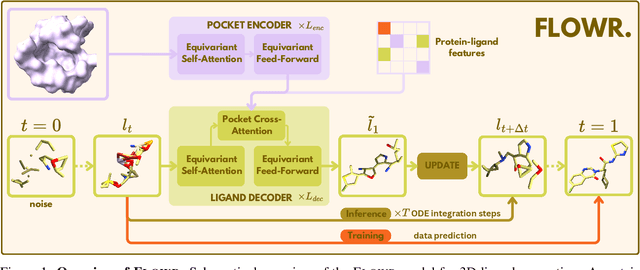
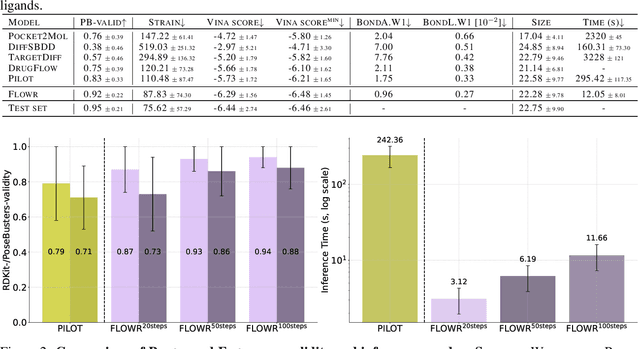
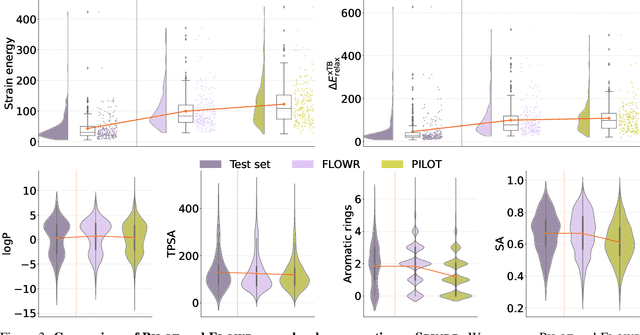
We introduce FLOWR, a novel structure-based framework for the generation and optimization of three-dimensional ligands. FLOWR integrates continuous and categorical flow matching with equivariant optimal transport, enhanced by an efficient protein pocket conditioning. Alongside FLOWR, we present SPINDR, a thoroughly curated dataset comprising ligand-pocket co-crystal complexes specifically designed to address existing data quality issues. Empirical evaluations demonstrate that FLOWR surpasses current state-of-the-art diffusion- and flow-based methods in terms of PoseBusters-validity, pose accuracy, and interaction recovery, while offering a significant inference speedup, achieving up to 70-fold faster performance. In addition, we introduce FLOWR.multi, a highly accurate multi-purpose model allowing for the targeted sampling of novel ligands that adhere to predefined interaction profiles and chemical substructures for fragment-based design without the need of re-training or any re-sampling strategies
Generative Modeling on Lie Groups via Euclidean Generalized Score Matching
Feb 04, 2025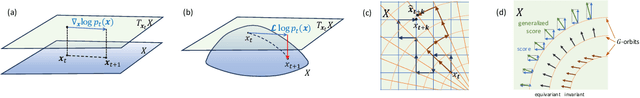
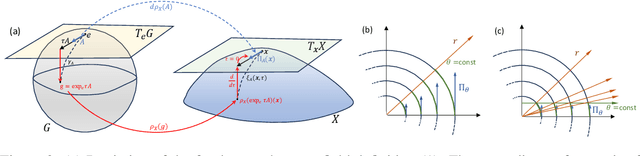

We extend Euclidean score-based diffusion processes to generative modeling on Lie groups. Through the formalism of Generalized Score Matching, our approach yields a Langevin dynamics which decomposes as a direct sum of Lie algebra representations, enabling generative processes on Lie groups while operating in Euclidean space. Unlike equivariant models, which restrict the space of learnable functions by quotienting out group orbits, our method can model any target distribution on any (non-Abelian) Lie group. Standard score matching emerges as a special case of our framework when the Lie group is the translation group. We prove that our generalized generative processes arise as solutions to a new class of paired stochastic differential equations (SDEs), introduced here for the first time. We validate our approach through experiments on diverse data types, demonstrating its effectiveness in real-world applications such as SO(3)-guided molecular conformer generation and modeling ligand-specific global SE(3) transformations for molecular docking, showing improvement in comparison to Riemannian diffusion on the group itself. We show that an appropriate choice of Lie group enhances learning efficiency by reducing the effective dimensionality of the trajectory space and enables the modeling of transitions between complex data distributions. Additionally, we demonstrate the universality of our approach by deriving how it extends to flow matching.
Knowledge Graph Based Agent for Complex, Knowledge-Intensive QA in Medicine
Oct 07, 2024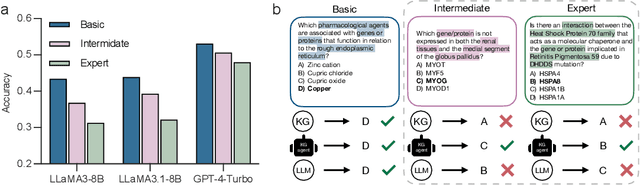
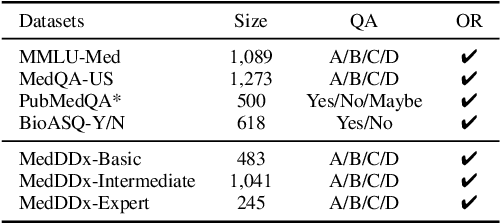
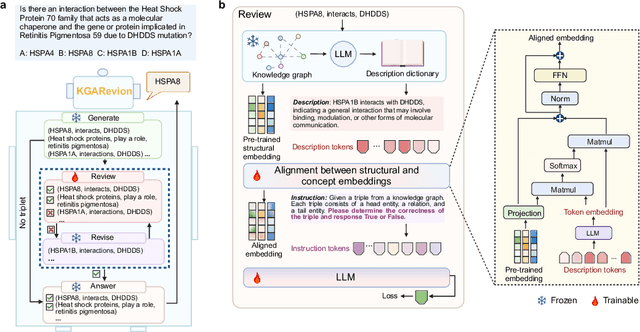
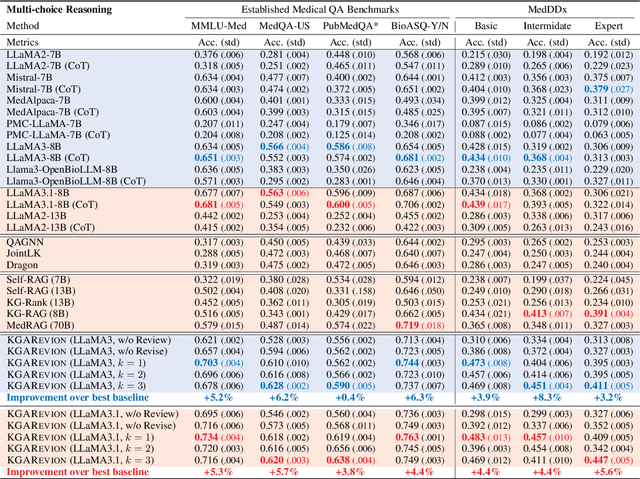
Biomedical knowledge is uniquely complex and structured, requiring distinct reasoning strategies compared to other scientific disciplines like physics or chemistry. Biomedical scientists do not rely on a single approach to reasoning; instead, they use various strategies, including rule-based, prototype-based, and case-based reasoning. This diversity calls for flexible approaches that accommodate multiple reasoning strategies while leveraging in-domain knowledge. We introduce KGARevion, a knowledge graph (KG) based agent designed to address the complexity of knowledge-intensive medical queries. Upon receiving a query, KGARevion generates relevant triplets by using the knowledge base of the LLM. These triplets are then verified against a grounded KG to filter out erroneous information and ensure that only accurate, relevant data contribute to the final answer. Unlike RAG-based models, this multi-step process ensures robustness in reasoning while adapting to different models of medical reasoning. Evaluations on four gold-standard medical QA datasets show that KGARevion improves accuracy by over 5.2%, outperforming 15 models in handling complex medical questions. To test its capabilities, we curated three new medical QA datasets with varying levels of semantic complexity, where KGARevion achieved a 10.4% improvement in accuracy.
PILOT: Equivariant diffusion for pocket conditioned de novo ligand generation with multi-objective guidance via importance sampling
May 23, 2024



The generation of ligands that both are tailored to a given protein pocket and exhibit a range of desired chemical properties is a major challenge in structure-based drug design. Here, we propose an in-silico approach for the $\textit{de novo}$ generation of 3D ligand structures using the equivariant diffusion model PILOT, combining pocket conditioning with a large-scale pre-training and property guidance. Its multi-objective trajectory-based importance sampling strategy is designed to direct the model towards molecules that not only exhibit desired characteristics such as increased binding affinity for a given protein pocket but also maintains high synthetic accessibility. This ensures the practicality of sampled molecules, thus maximizing their potential for the drug discovery pipeline. PILOT significantly outperforms existing methods across various metrics on the common benchmark dataset CrossDocked2020. Moreover, we employ PILOT to generate novel ligands for unseen protein pockets from the Kinodata-3D dataset, which encompasses a substantial portion of the human kinome. The generated structures exhibit predicted $IC_{50}$ values indicative of potent biological activity, which highlights the potential of PILOT as a powerful tool for structure-based drug design.
Navigating the Design Space of Equivariant Diffusion-Based Generative Models for De Novo 3D Molecule Generation
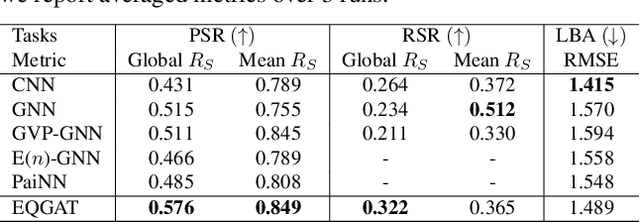
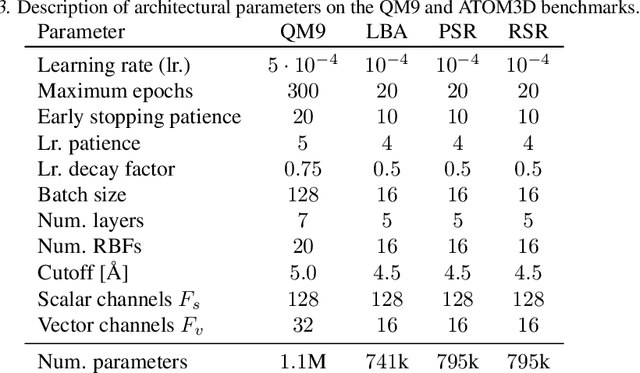
Sep 29, 2023Deep generative diffusion models are a promising avenue for de novo 3D molecular design in material science and drug discovery. However, their utility is still constrained by suboptimal performance with large molecular structures and limited training data. Addressing this gap, we explore the design space of E(3) equivariant diffusion models, focusing on previously blank spots. Our extensive comparative analysis evaluates the interplay between continuous and discrete state spaces. Out of this investigation, we introduce the EQGAT-diff model, which consistently surpasses the performance of established models on the QM9 and GEOM-Drugs datasets by a large margin. Distinctively, EQGAT-diff takes continuous atomic positions while chemical elements and bond types are categorical and employ a time-dependent loss weighting that significantly increases training convergence and the quality of generated samples. To further strengthen the applicability of diffusion models to limited training data, we examine the transferability of EQGAT-diff trained on the large PubChem3D dataset with implicit hydrogens to target distributions with explicit hydrogens. Fine-tuning EQGAT-diff for a couple of iterations further pushes state-of-the-art performance across datasets. We envision that our findings will find applications in structure-based drug design, where the accuracy of generative models for small datasets of complex molecules is critical.
From slides (through tiles) to pixels: an explainability framework for weakly supervised models in pre-clinical pathology
Feb 03, 2023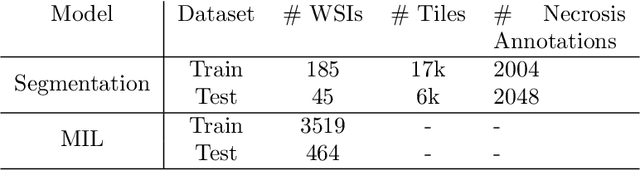
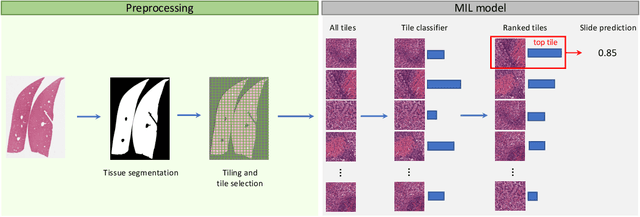
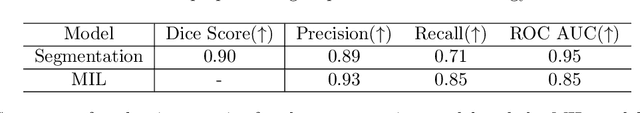
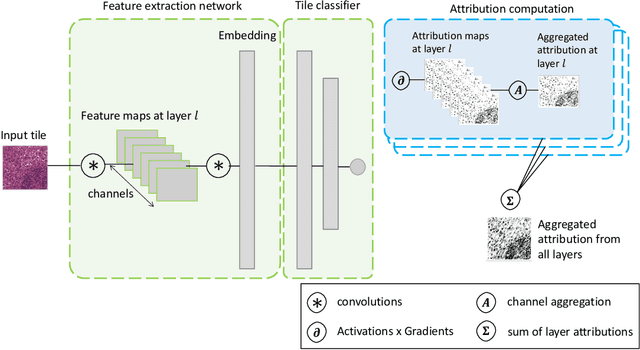
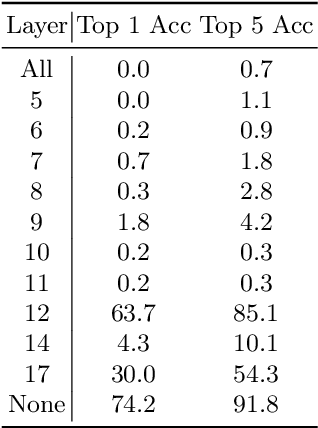
In pre-clinical pathology, there is a paradox between the abundance of raw data (whole slide images from many organs of many individual animals) and the lack of pixel-level slide annotations done by pathologists. Due to time constraints and requirements from regulatory authorities, diagnoses are instead stored as slide labels. Weakly supervised training is designed to take advantage of those data, and the trained models can be used by pathologists to rank slides by their probability of containing a given lesion of interest. In this work, we propose a novel contextualized eXplainable AI (XAI) framework and its application to deep learning models trained on Whole Slide Images (WSIs) in Digital Pathology. Specifically, we apply our methods to a multi-instance-learning (MIL) model, which is trained solely on slide-level labels, without the need for pixel-level annotations. We validate quantitatively our methods by quantifying the agreements of our explanations' heatmaps with pathologists' annotations, as well as with predictions from a segmentation model trained on such annotations. We demonstrate the stability of the explanations with respect to input shifts, and the fidelity with respect to increased model performance. We quantitatively evaluate the correlation between available pixel-wise annotations and explainability heatmaps. We show that the explanations on important tiles of the whole slide correlate with tissue changes between healthy regions and lesions, but do not exactly behave like a human annotator. This result is coherent with the model training strategy.
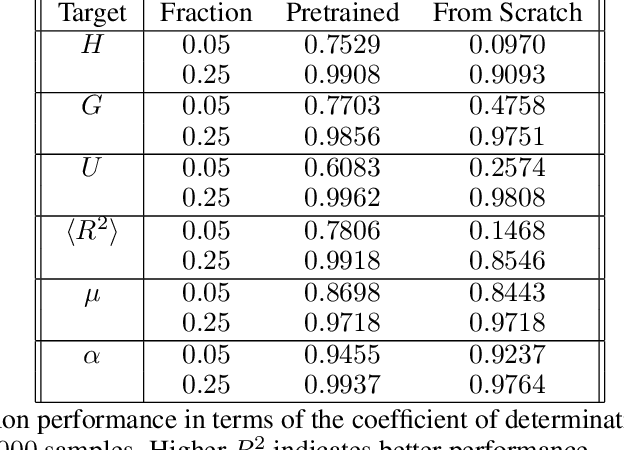
Equivariant Graph Attention Networks for Molecular Property Prediction
Mar 02, 2022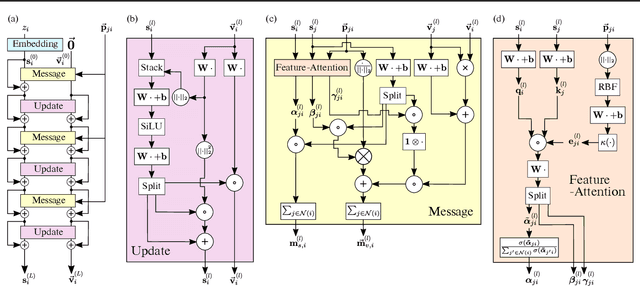
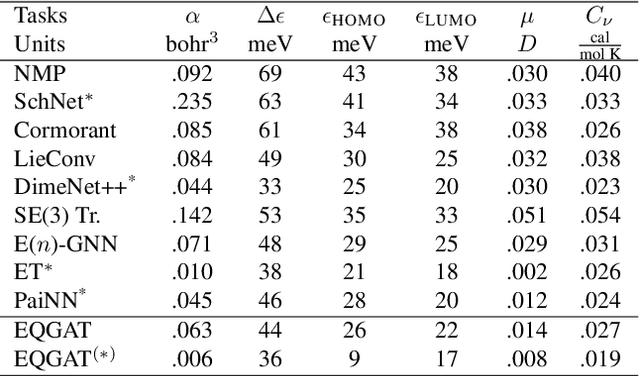
Learning and reasoning about 3D molecular structures with varying size is an emerging and important challenge in machine learning and especially in drug discovery. Equivariant Graph Neural Networks (GNNs) can simultaneously leverage the geometric and relational detail of the problem domain and are known to learn expressive representations through the propagation of information between nodes leveraging higher-order representations to faithfully express the geometry of the data, such as directionality in their intermediate layers. In this work, we propose an equivariant GNN that operates with Cartesian coordinates to incorporate directionality and we implement a novel attention mechanism, acting as a content and spatial dependent filter when propagating information between nodes. We demonstrate the efficacy of our architecture on predicting quantum mechanical properties of small molecules and its benefit on problems that concern macromolecular structures such as protein complexes.
Explaining, Evaluating and Enhancing Neural Networks' Learned Representations
Feb 18, 2022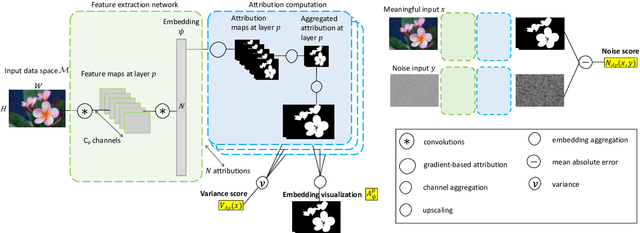
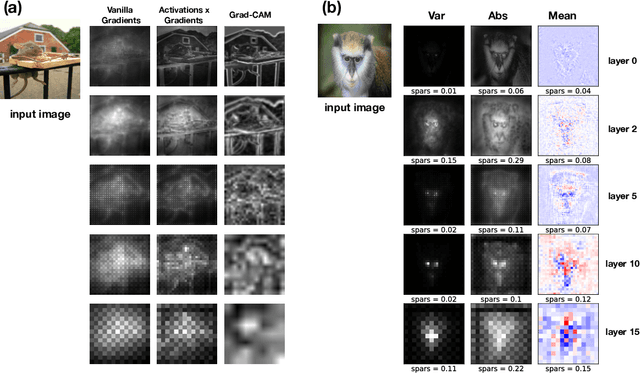
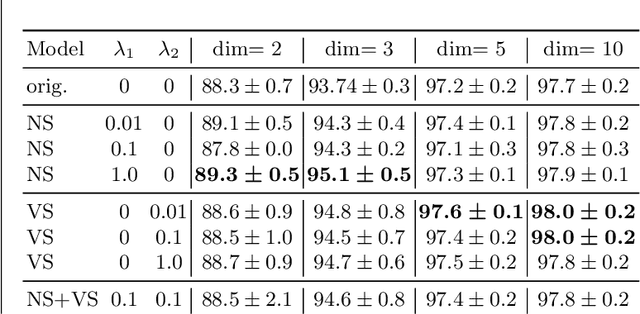
Most efforts in interpretability in deep learning have focused on (1) extracting explanations of a specific downstream task in relation to the input features and (2) imposing constraints on the model, often at the expense of predictive performance. New advances in (unsupervised) representation learning and transfer learning, however, raise the need for an explanatory framework for networks that are trained without a specific downstream task. We address these challenges by showing how explainability can be an aid, rather than an obstacle, towards better and more efficient representations. Specifically, we propose a natural aggregation method generalizing attribution maps between any two (convolutional) layers of a neural network. Additionally, we employ such attributions to define two novel scores for evaluating the informativeness and the disentanglement of latent embeddings. Extensive experiments show that the proposed scores do correlate with the desired properties. We also confirm and extend previously known results concerning the independence of some common saliency strategies from the model parameters. Finally, we show that adopting our proposed scores as constraints during the training of a representation learning task improves the downstream performance of the model.
Unsupervised Learning of Group Invariant and Equivariant Representations
Feb 15, 2022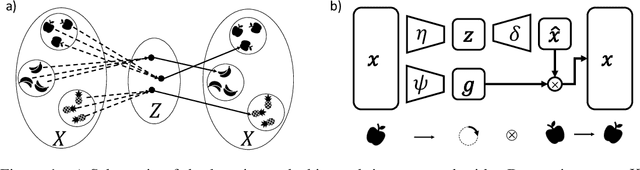
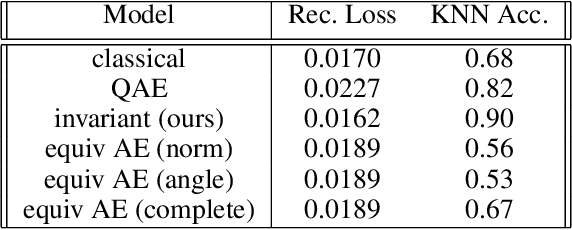

Equivariant neural networks, whose hidden features transform according to representations of a group G acting on the data, exhibit training efficiency and an improved generalisation performance. In this work, we extend group invariant and equivariant representation learning to the field of unsupervised deep learning. We propose a general learning strategy based on an encoder-decoder framework in which the latent representation is disentangled in an invariant term and an equivariant group action component. The key idea is that the network learns the group action on the data space and thus is able to solve the reconstruction task from an invariant data representation, hence avoiding the necessity of ad-hoc group-specific implementations. We derive the necessary conditions on the equivariant encoder, and we present a construction valid for any G, both discrete and continuous. We describe explicitly our construction for rotations, translations and permutations. We test the validity and the robustness of our approach in a variety of experiments with diverse data types employing different network architectures.