 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAssessing generative modeling approaches for free energy estimates in condensed matter
Dec 30, 2025The accurate estimation of free energy differences between two states is a long-standing challenge in molecular simulations. Traditional approaches generally rely on sampling multiple intermediate states to ensure sufficient overlap in phase space and are, consequently, computationally expensive. Several generative-model-based methods have recently addressed this challenge by learning a direct bridge between distributions, bypassing the need for intermediate states. However, it remains unclear which approaches provide the best trade-off between efficiency, accuracy, and scalability. In this work, we systematically review these methods and benchmark selected approaches with a focus on condensed-matter systems. In particular, we investigate the performance of discrete and continuous normalizing flows in the context of targeted free energy perturbation as well as FEAT (Free energy Estimators with Adaptive Transport) together with the escorted Jarzynski equality, using coarse-grained monatomic ice and Lennard-Jones solids as benchmark systems. We evaluate accuracy, data efficiency, computational cost, and scalability with system size. Our results provide a quantitative framework for selecting effective free energy estimation strategies in condensed-phase systems.
Estimating Solvation Free Energies with Boltzmann Generators
Dec 20, 2025


Accurate calculations of solvation free energies remain a central challenge in molecular simulations, often requiring extensive sampling and numerous alchemical intermediates to ensure sufficient overlap between phase-space distributions of a solute in the gas phase and in solution. Here, we introduce a computational framework based on normalizing flows that directly maps solvent configurations between solutes of different sizes, and compare the accuracy and efficiency to conventional free energy estimates. For a Lennard-Jones solvent, we demonstrate that this approach yields acceptable accuracy in estimating free energy differences for challenging transformations, such as solute growth or increased solute-solute separation, which typically demand multiple intermediate simulation steps along the transformation. Analysis of radial distribution functions indicates that the flow generates physically meaningful solvent rearrangements, substantially enhancing configurational overlap between states in configuration space. These results suggest flow-based models as a promising alternative to traditional free energy estimation methods.
Efficient mapping of phase diagrams with conditional normalizing flows
Jun 18, 2024The accurate prediction of phase diagrams is of central importance for both the fundamental understanding of materials as well as for technological applications in material sciences. However, the computational prediction of the relative stability between phases based on their free energy is a daunting task, as traditional free energy estimators require a large amount of simulation data to obtain uncorrelated equilibrium samples over a grid of thermodynamic states. In this work, we develop deep generative machine learning models for entire phase diagrams, employing normalizing flows conditioned on the thermodynamic states, e.g., temperature and pressure, that they map to. By training a single normalizing flow to transform the equilibrium distribution sampled at only one reference thermodynamic state to a wide range of target temperatures and pressures, we can efficiently generate equilibrium samples across the entire phase diagram. Using a permutation-equivariant architecture allows us, thereby, to treat solid and liquid phases on the same footing. We demonstrate our approach by predicting the solid-liquid coexistence line for a Lennard-Jones system in excellent agreement with state-of-the-art free energy methods while significantly reducing the number of energy evaluations needed.
Multi-Type Point Cloud Autoencoder: A Complete Equivariant Embedding for Molecule Conformation and Pose
May 22, 2024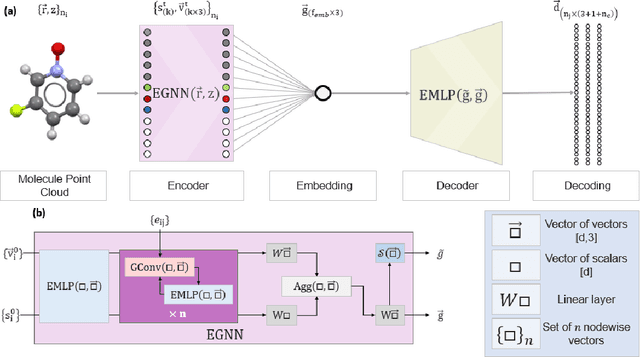
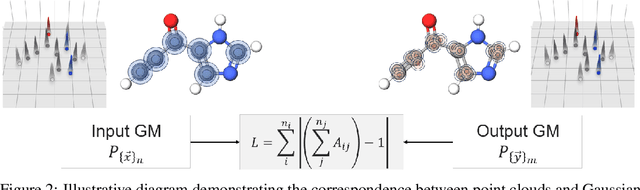
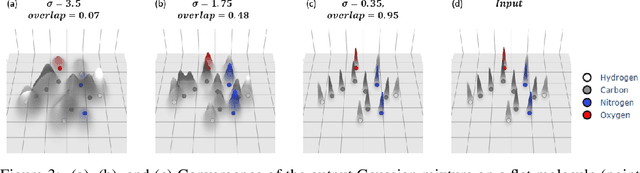
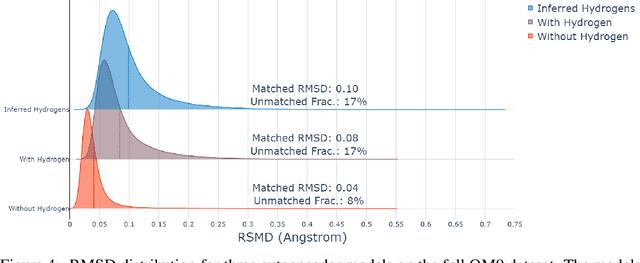
The point cloud is a flexible representation for a wide variety of data types, and is a particularly natural fit for the 3D conformations of molecules. Extant molecule embedding/representation schemes typically focus on internal degrees of freedom, ignoring the global 3D orientation. For tasks that depend on knowledge of both molecular conformation and 3D orientation, such as the generation of molecular dimers, clusters, or condensed phases, we require a representation which is provably complete in the types and positions of atomic nuclei and roto-inversion equivariant with respect to the input point cloud. We develop, train, and evaluate a new type of autoencoder, molecular O(3) encoding net (Mo3ENet), for multi-type point clouds, for which we propose a new reconstruction loss, capitalizing on a Gaussian mixture representation of the input and output point clouds. Mo3ENet is end-to-end equivariant, meaning the learned representation can be manipulated on O(3), a practical bonus for downstream learning tasks. An appropriately trained Mo3ENet latent space comprises a universal embedding for scalar and vector molecule property prediction tasks, as well as other downstream tasks incorporating the 3D molecular pose.
Geometric Deep Learning for Molecular Crystal Structure Prediction
Mar 17, 2023



We develop and test new machine learning strategies for accelerating molecular crystal structure ranking and crystal property prediction using tools from geometric deep learning on molecular graphs. Leveraging developments in graph-based learning and the availability of large molecular crystal datasets, we train models for density prediction and stability ranking which are accurate, fast to evaluate, and applicable to molecules of widely varying size and composition. Our density prediction model, MolXtalNet-D, achieves state of the art performance, with lower than 2% mean absolute error on a large and diverse test dataset. Our crystal ranking tool, MolXtalNet-S, correctly discriminates experimental samples from synthetically generated fakes and is further validated through analysis of the submissions to the Cambridge Structural Database Blind Tests 5 and 6. Our new tools are computationally cheap and flexible enough to be deployed within an existing crystal structure prediction pipeline both to reduce the search space and score/filter crystal candidates.