 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMulti-Type Point Cloud Autoencoder: A Complete Equivariant Embedding for Molecule Conformation and Pose
May 22, 2024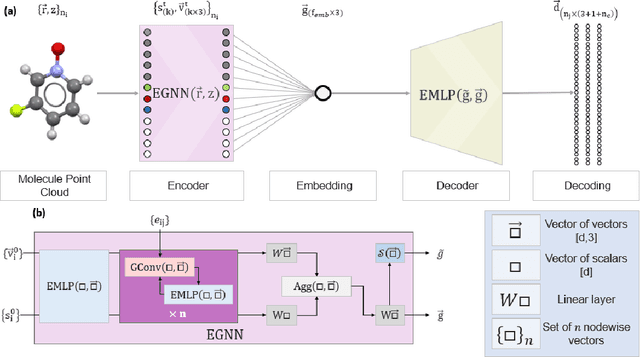
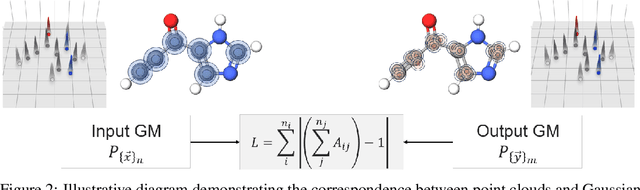
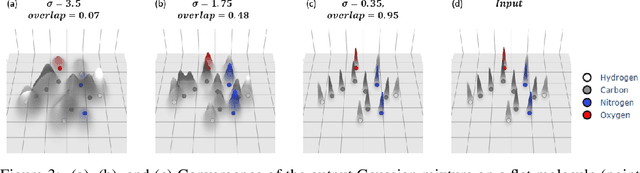
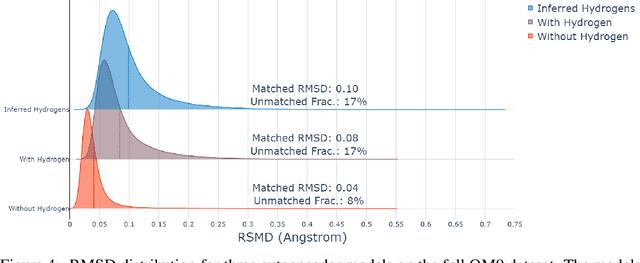
The point cloud is a flexible representation for a wide variety of data types, and is a particularly natural fit for the 3D conformations of molecules. Extant molecule embedding/representation schemes typically focus on internal degrees of freedom, ignoring the global 3D orientation. For tasks that depend on knowledge of both molecular conformation and 3D orientation, such as the generation of molecular dimers, clusters, or condensed phases, we require a representation which is provably complete in the types and positions of atomic nuclei and roto-inversion equivariant with respect to the input point cloud. We develop, train, and evaluate a new type of autoencoder, molecular O(3) encoding net (Mo3ENet), for multi-type point clouds, for which we propose a new reconstruction loss, capitalizing on a Gaussian mixture representation of the input and output point clouds. Mo3ENet is end-to-end equivariant, meaning the learned representation can be manipulated on O(3), a practical bonus for downstream learning tasks. An appropriately trained Mo3ENet latent space comprises a universal embedding for scalar and vector molecule property prediction tasks, as well as other downstream tasks incorporating the 3D molecular pose.
Geometric Deep Learning for Molecular Crystal Structure Prediction
Mar 17, 2023



We develop and test new machine learning strategies for accelerating molecular crystal structure ranking and crystal property prediction using tools from geometric deep learning on molecular graphs. Leveraging developments in graph-based learning and the availability of large molecular crystal datasets, we train models for density prediction and stability ranking which are accurate, fast to evaluate, and applicable to molecules of widely varying size and composition. Our density prediction model, MolXtalNet-D, achieves state of the art performance, with lower than 2% mean absolute error on a large and diverse test dataset. Our crystal ranking tool, MolXtalNet-S, correctly discriminates experimental samples from synthetically generated fakes and is further validated through analysis of the submissions to the Cambridge Structural Database Blind Tests 5 and 6. Our new tools are computationally cheap and flexible enough to be deployed within an existing crystal structure prediction pipeline both to reduce the search space and score/filter crystal candidates.
Diversifying Design of Nucleic Acid Aptamers Using Unsupervised Machine Learning
Aug 10, 2022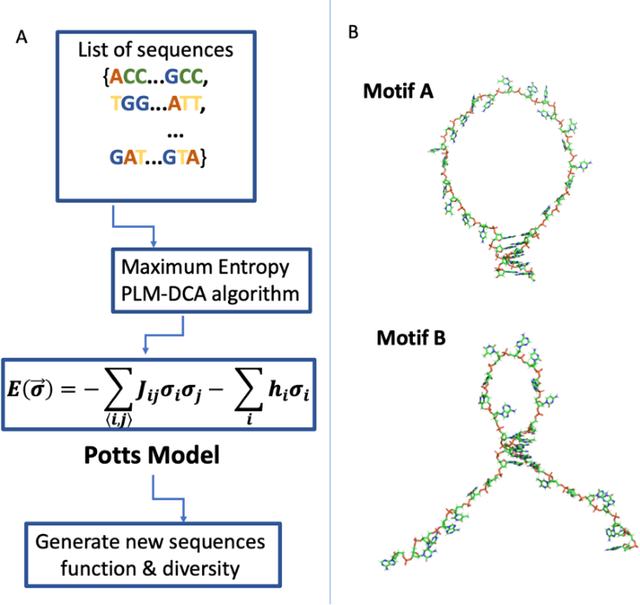
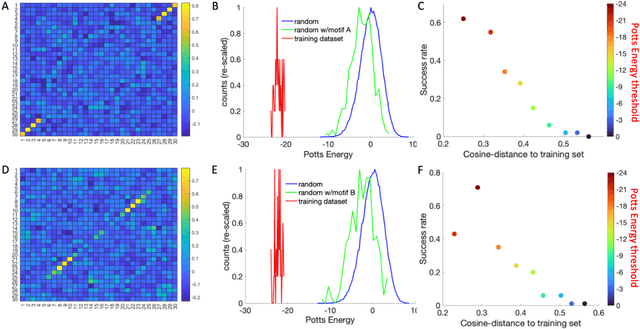
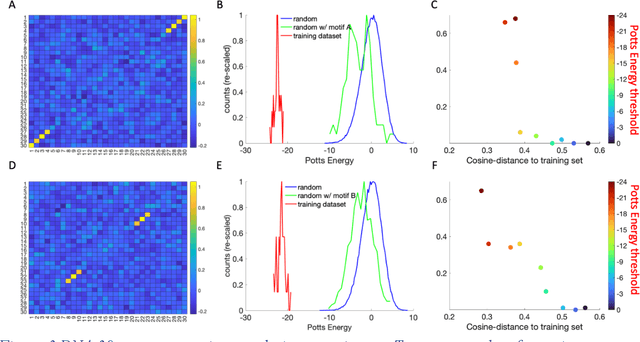
Inverse design of short single-stranded RNA and DNA sequences (aptamers) is the task of finding sequences that satisfy a set of desired criteria. Relevant criteria may be, for example, the presence of specific folding motifs, binding to molecular ligands, sensing properties, etc. Most practical approaches to aptamer design identify a small set of promising candidate sequences using high-throughput experiments (e.g. SELEX), and then optimize performance by introducing only minor modifications to the empirically found candidates. Sequences that possess the desired properties but differ drastically in chemical composition will add diversity to the search space and facilitate the discovery of useful nucleic acid aptamers. Systematic diversification protocols are needed. Here we propose to use an unsupervised machine learning model known as the Potts model to discover new, useful sequences with controllable sequence diversity. We start by training a Potts model using the maximum entropy principle on a small set of empirically identified sequences unified by a common feature. To generate new candidate sequences with a controllable degree of diversity, we take advantage of the model's spectral feature: an energy bandgap separating sequences that are similar to the training set from those that are distinct. By controlling the Potts energy range that is sampled, we generate sequences that are distinct from the training set yet still likely to have the encoded features. To demonstrate performance, we apply our approach to design diverse pools of sequences with specified secondary structure motifs in 30-mer RNA and DNA aptamers.