 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEfficient Evolutionary Search Over Chemical Space with Large Language Models
Jun 23, 2024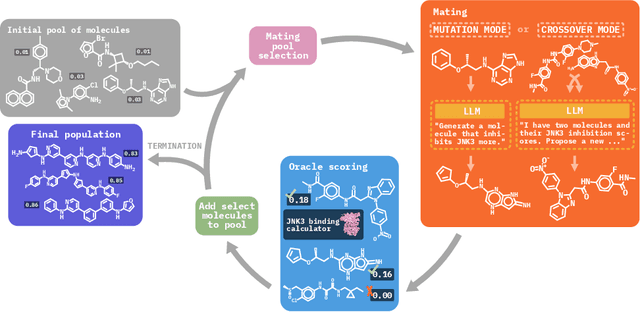
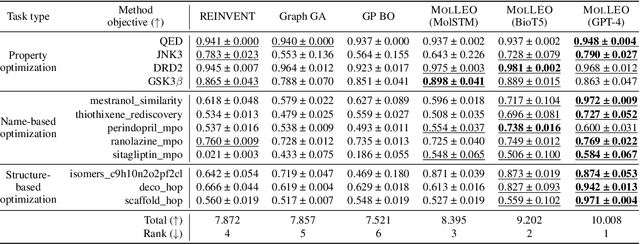
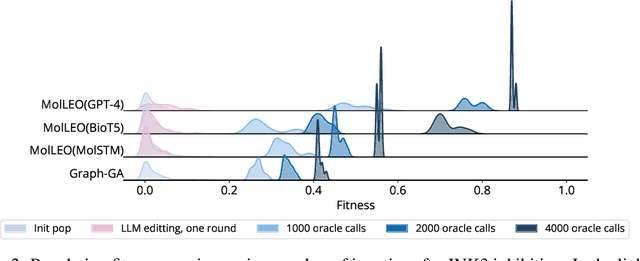
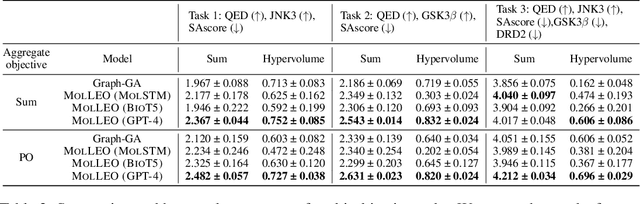
Molecular discovery, when formulated as an optimization problem, presents significant computational challenges because optimization objectives can be non-differentiable. Evolutionary Algorithms (EAs), often used to optimize black-box objectives in molecular discovery, traverse chemical space by performing random mutations and crossovers, leading to a large number of expensive objective evaluations. In this work, we ameliorate this shortcoming by incorporating chemistry-aware Large Language Models (LLMs) into EAs. Namely, we redesign crossover and mutation operations in EAs using LLMs trained on large corpora of chemical information. We perform extensive empirical studies on both commercial and open-source models on multiple tasks involving property optimization, molecular rediscovery, and structure-based drug design, demonstrating that the joint usage of LLMs with EAs yields superior performance over all baseline models across single- and multi-objective settings. We demonstrate that our algorithm improves both the quality of the final solution and convergence speed, thereby reducing the number of required objective evaluations. Our code is available at http://github.com/zoom-wang112358/MOLLEO