 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRetroDiff: Retrosynthesis as Multi-stage Distribution Interpolation
Nov 23, 2023Retrosynthesis poses a fundamental challenge in biopharmaceuticals, aiming to aid chemists in finding appropriate reactant molecules and synthetic pathways given determined product molecules. With the reactant and product represented as 2D graphs, retrosynthesis constitutes a conditional graph-to-graph generative task. Inspired by the recent advancements in discrete diffusion models for graph generation, we introduce Retrosynthesis Diffusion (RetroDiff), a novel diffusion-based method designed to address this problem. However, integrating a diffusion-based graph-to-graph framework while retaining essential chemical reaction template information presents a notable challenge. Our key innovation is to develop a multi-stage diffusion process. In this method, we decompose the retrosynthesis procedure to first sample external groups from the dummy distribution given products and then generate the external bonds to connect the products and generated groups. Interestingly, such a generation process is exactly the reverse of the widely adapted semi-template retrosynthesis procedure, i.e. from reaction center identification to synthon completion, which significantly reduces the error accumulation. Experimental results on the benchmark have demonstrated the superiority of our method over all other semi-template methods.
Protein-ligand binding representation learning from fine-grained interactions
Nov 09, 2023



The binding between proteins and ligands plays a crucial role in the realm of drug discovery. Previous deep learning approaches have shown promising results over traditional computationally intensive methods, but resulting in poor generalization due to limited supervised data. In this paper, we propose to learn protein-ligand binding representation in a self-supervised learning manner. Different from existing pre-training approaches which treat proteins and ligands individually, we emphasize to discern the intricate binding patterns from fine-grained interactions. Specifically, this self-supervised learning problem is formulated as a prediction of the conclusive binding complex structure given a pocket and ligand with a Transformer based interaction module, which naturally emulates the binding process. To ensure the representation of rich binding information, we introduce two pre-training tasks, i.e.~atomic pairwise distance map prediction and mask ligand reconstruction, which comprehensively model the fine-grained interactions from both structure and feature space. Extensive experiments have demonstrated the superiority of our method across various binding tasks, including protein-ligand affinity prediction, virtual screening and protein-ligand docking.
UniMAP: Universal SMILES-Graph Representation Learning
Oct 22, 2023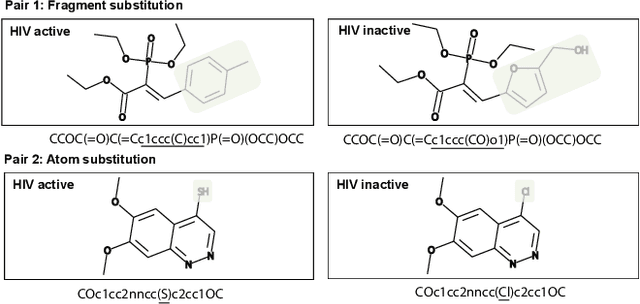
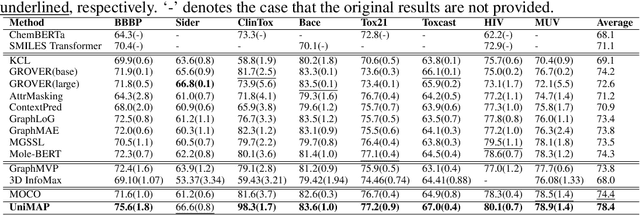
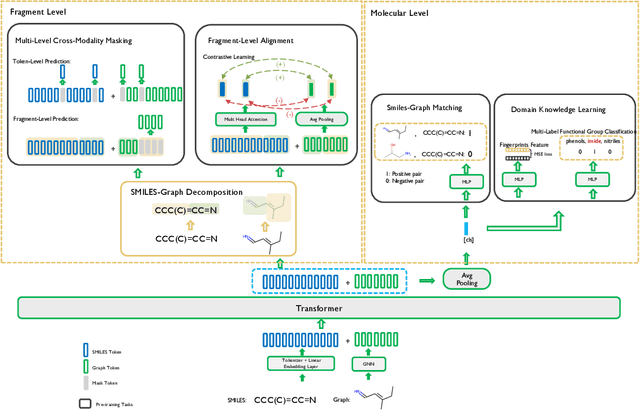
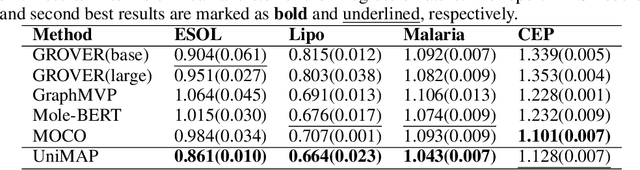
Molecular representation learning is fundamental for many drug related applications. Most existing molecular pre-training models are limited in using single molecular modality, either SMILES or graph representation. To effectively leverage both modalities, we argue that it is critical to capture the fine-grained 'semantics' between SMILES and graph, because subtle sequence/graph differences may lead to contrary molecular properties. In this paper, we propose a universal SMILE-graph representation learning model, namely UniMAP. Firstly, an embedding layer is employed to obtain the token and node/edge representation in SMILES and graph, respectively. A multi-layer Transformer is then utilized to conduct deep cross-modality fusion. Specially, four kinds of pre-training tasks are designed for UniMAP, including Multi-Level Cross-Modality Masking (CMM), SMILES-Graph Matching (SGM), Fragment-Level Alignment (FLA), and Domain Knowledge Learning (DKL). In this way, both global (i.e. SGM and DKL) and local (i.e. CMM and FLA) alignments are integrated to achieve comprehensive cross-modality fusion. We evaluate UniMAP on various downstream tasks, i.e. molecular property prediction, drug-target affinity prediction and drug-drug interaction. Experimental results show that UniMAP outperforms current state-of-the-art pre-training methods.We also visualize the learned representations to demonstrate the effect of multi-modality integration.
Self-supervised Pocket Pretraining via Protein Fragment-Surroundings Alignment
Oct 11, 2023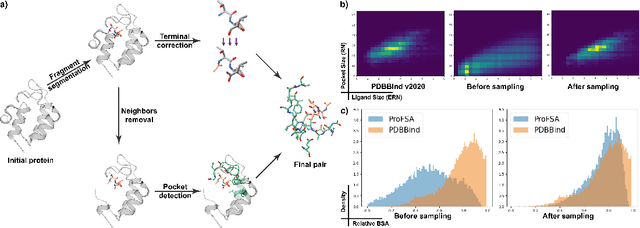
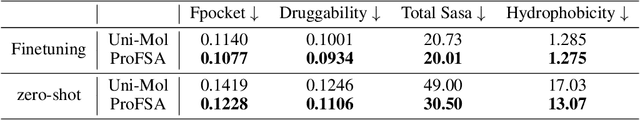
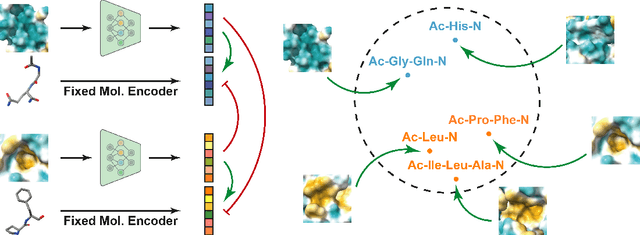

Pocket representations play a vital role in various biomedical applications, such as druggability estimation, ligand affinity prediction, and de novo drug design. While existing geometric features and pretrained representations have demonstrated promising results, they usually treat pockets independent of ligands, neglecting the fundamental interactions between them. However, the limited pocket-ligand complex structures available in the PDB database (less than 100 thousand non-redundant pairs) hampers large-scale pretraining endeavors for interaction modeling. To address this constraint, we propose a novel pocket pretraining approach that leverages knowledge from high-resolution atomic protein structures, assisted by highly effective pretrained small molecule representations. By segmenting protein structures into drug-like fragments and their corresponding pockets, we obtain a reasonable simulation of ligand-receptor interactions, resulting in the generation of over 5 million complexes. Subsequently, the pocket encoder is trained in a contrastive manner to align with the representation of pseudo-ligand furnished by some pretrained small molecule encoders. Our method, named ProFSA, achieves state-of-the-art performance across various tasks, including pocket druggability prediction, pocket matching, and ligand binding affinity prediction. Notably, ProFSA surpasses other pretraining methods by a substantial margin. Moreover, our work opens up a new avenue for mitigating the scarcity of protein-ligand complex data through the utilization of high-quality and diverse protein structure databases.
DrugCLIP: Contrastive Protein-Molecule Representation Learning for Virtual Screening
Oct 10, 2023Virtual screening, which identifies potential drugs from vast compound databases to bind with a particular protein pocket, is a critical step in AI-assisted drug discovery. Traditional docking methods are highly time-consuming, and can only work with a restricted search library in real-life applications. Recent supervised learning approaches using scoring functions for binding-affinity prediction, although promising, have not yet surpassed docking methods due to their strong dependency on limited data with reliable binding-affinity labels. In this paper, we propose a novel contrastive learning framework, DrugCLIP, by reformulating virtual screening as a dense retrieval task and employing contrastive learning to align representations of binding protein pockets and molecules from a large quantity of pairwise data without explicit binding-affinity scores. We also introduce a biological-knowledge inspired data augmentation strategy to learn better protein-molecule representations. Extensive experiments show that DrugCLIP significantly outperforms traditional docking and supervised learning methods on diverse virtual screening benchmarks with highly reduced computation time, especially in zero-shot setting.