 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMulti-level Interaction Modeling for Protein Mutational Effect Prediction
May 28, 2024



Protein-protein interactions are central mediators in many biological processes. Accurately predicting the effects of mutations on interactions is crucial for guiding the modulation of these interactions, thereby playing a significant role in therapeutic development and drug discovery. Mutations generally affect interactions hierarchically across three levels: mutated residues exhibit different sidechain conformations, which lead to changes in the backbone conformation, eventually affecting the binding affinity between proteins. However, existing methods typically focus only on sidechain-level interaction modeling, resulting in suboptimal predictions. In this work, we propose a self-supervised multi-level pre-training framework, ProMIM, to fully capture all three levels of interactions with well-designed pretraining objectives. Experiments show ProMIM outperforms all the baselines on the standard benchmark, especially on mutations where significant changes in backbone conformations may occur. In addition, leading results from zero-shot evaluations for SARS-CoV-2 mutational effect prediction and antibody optimization underscore the potential of ProMIM as a powerful next-generation tool for developing novel therapeutic approaches and new drugs.
Self-supervised Pocket Pretraining via Protein Fragment-Surroundings Alignment
Oct 11, 2023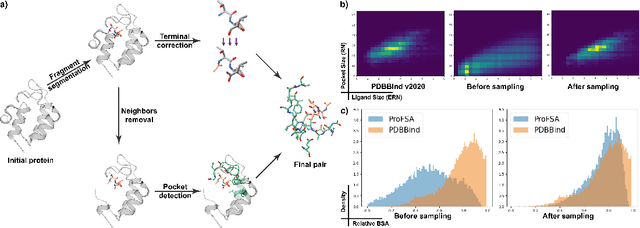
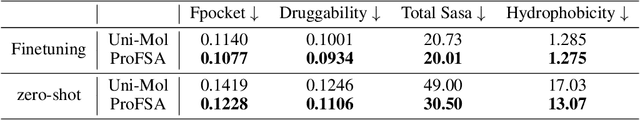
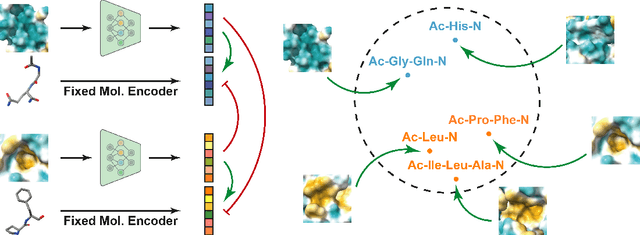

Pocket representations play a vital role in various biomedical applications, such as druggability estimation, ligand affinity prediction, and de novo drug design. While existing geometric features and pretrained representations have demonstrated promising results, they usually treat pockets independent of ligands, neglecting the fundamental interactions between them. However, the limited pocket-ligand complex structures available in the PDB database (less than 100 thousand non-redundant pairs) hampers large-scale pretraining endeavors for interaction modeling. To address this constraint, we propose a novel pocket pretraining approach that leverages knowledge from high-resolution atomic protein structures, assisted by highly effective pretrained small molecule representations. By segmenting protein structures into drug-like fragments and their corresponding pockets, we obtain a reasonable simulation of ligand-receptor interactions, resulting in the generation of over 5 million complexes. Subsequently, the pocket encoder is trained in a contrastive manner to align with the representation of pseudo-ligand furnished by some pretrained small molecule encoders. Our method, named ProFSA, achieves state-of-the-art performance across various tasks, including pocket druggability prediction, pocket matching, and ligand binding affinity prediction. Notably, ProFSA surpasses other pretraining methods by a substantial margin. Moreover, our work opens up a new avenue for mitigating the scarcity of protein-ligand complex data through the utilization of high-quality and diverse protein structure databases.