 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFrom Theory to Therapy: Reframing SBDD Model Evaluation via Practical Metrics
Jun 13, 2024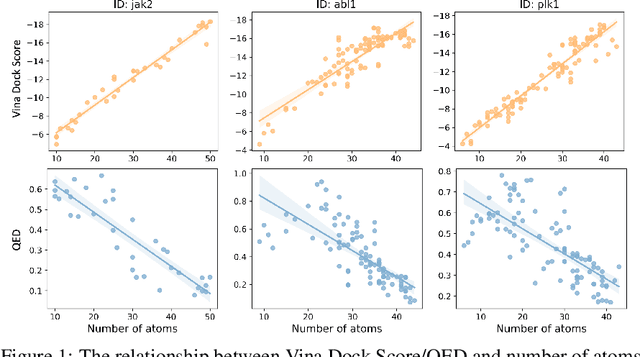
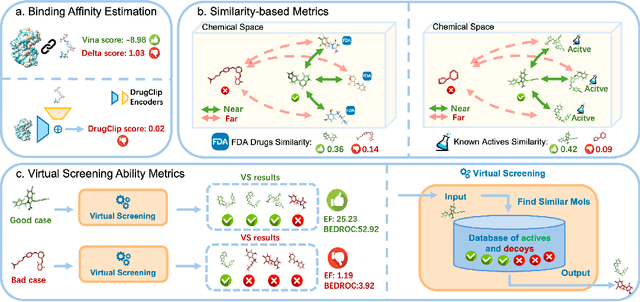
Recent advancements in structure-based drug design (SBDD) have significantly enhanced the efficiency and precision of drug discovery by generating molecules tailored to bind specific protein pockets. Despite these technological strides, their practical application in real-world drug development remains challenging due to the complexities of synthesizing and testing these molecules. The reliability of the Vina docking score, the current standard for assessing binding abilities, is increasingly questioned due to its susceptibility to overfitting. To address these limitations, we propose a comprehensive evaluation framework that includes assessing the similarity of generated molecules to known active compounds, introducing a virtual screening-based metric for practical deployment capabilities, and re-evaluating binding affinity more rigorously. Our experiments reveal that while current SBDD models achieve high Vina scores, they fall short in practical usability metrics, highlighting a significant gap between theoretical predictions and real-world applicability. Our proposed metrics and dataset aim to bridge this gap, enhancing the practical applicability of future SBDD models and aligning them more closely with the needs of pharmaceutical research and development.
Delta Score: Improving the Binding Assessment of Structure-Based Drug Design Methods
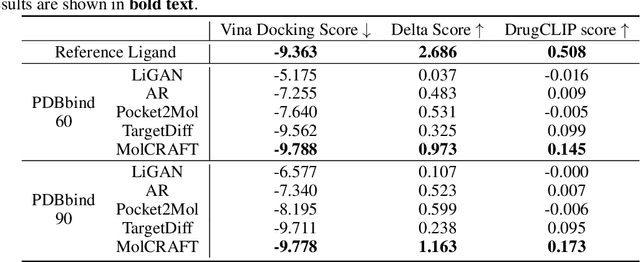
Nov 01, 2023Structure-based drug design (SBDD) stands at the forefront of drug discovery, emphasizing the creation of molecules that target specific binding pockets. Recent advances in this area have witnessed the adoption of deep generative models and geometric deep learning techniques, modeling SBDD as a conditional generation task where the target structure serves as context. Historically, evaluation of these models centered on docking scores, which quantitatively depict the predicted binding affinity between a molecule and its target pocket. Though state-of-the-art models purport that a majority of their generated ligands exceed the docking score of ground truth ligands in test sets, it begs the question: Do these scores align with real-world biological needs? In this paper, we introduce the delta score, a novel evaluation metric grounded in tangible pharmaceutical requisites. Our experiments reveal that molecules produced by current deep generative models significantly lag behind ground truth reference ligands when assessed with the delta score. This novel metric not only complements existing benchmarks but also provides a pivotal direction for subsequent research in the domain.
DrugCLIP: Contrastive Protein-Molecule Representation Learning for Virtual Screening
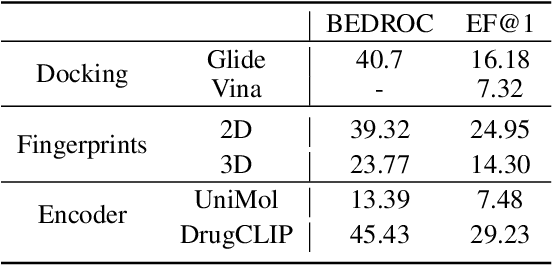
Oct 10, 2023Virtual screening, which identifies potential drugs from vast compound databases to bind with a particular protein pocket, is a critical step in AI-assisted drug discovery. Traditional docking methods are highly time-consuming, and can only work with a restricted search library in real-life applications. Recent supervised learning approaches using scoring functions for binding-affinity prediction, although promising, have not yet surpassed docking methods due to their strong dependency on limited data with reliable binding-affinity labels. In this paper, we propose a novel contrastive learning framework, DrugCLIP, by reformulating virtual screening as a dense retrieval task and employing contrastive learning to align representations of binding protein pockets and molecules from a large quantity of pairwise data without explicit binding-affinity scores. We also introduce a biological-knowledge inspired data augmentation strategy to learn better protein-molecule representations. Extensive experiments show that DrugCLIP significantly outperforms traditional docking and supervised learning methods on diverse virtual screening benchmarks with highly reduced computation time, especially in zero-shot setting.