 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDemystifying Multimodal Biomolecular Co-design With Intrinsic Geodesic Coupling
Jun 01, 2026Biomolecules such as proteins and small-molecule ligands play a central role in biological systems, arising from the tight interplay between sequence and three-dimensional structure. Recent generative models for biomolecular co-design aim to capture this interplay by jointly modeling coupled modalities. However, existing approaches largely adopt a parallel execution of marginal generative processes, implicitly enforcing fixed synchronous coupling. We argue that a critical but overlooked degree of freedom lies in how these marginal processes are temporally coupled during training and generation, where inappropriate coupling can introduce high-variance supervision and inconsistent intermediate states, affecting modality consistency. To address this, we introduce GeoCoupling, a systematic framework that optimizes for temporal couplings between heterogeneous modalities. Empirical results across structure-based drug design and unconditional protein design demonstrate the learned couplings consistently outperform synchronous and randomly coupled baselines, yielding biomolecules with improved physical validity and diversity.
AMix-2: Establishing Protein as a Native Modality in Large Language Models
May 29, 2026We present AMix-2, a protein-text foundation model that establishes protein as a native modality in large language models (LLMs), unifying protein understanding and sequence design within a single foundation model. AMix-2 is built upon two key ideas: (1) a unified protein-text formulation that embeds natural language and protein sequence in a shared token space, enabling one model to perform biological reasoning and conditional design instead of separate downstream task-specialized models; and (2) a block-wise diffusion language modeling backbone that combines causal generation across blocks with bidirectional context and iterative refinement within blocks. This scheme better matches the intrinsic nature of proteins than a strict left-to-right factorization. To evaluate protein foundation models under realistic generalization settings, we further introduce ProteinArena, a comprehensive benchmark with time-aware and homology-aware protocols across various understanding and design tasks, and with baselines covering classical bioinformatics tools, protein-specialized models and LLMs. On ProteinArena, AMix-2 outperforms frontier LLMs and demonstrates competitive performance to task-specific protein models. Controlled experiments further show that the diffusion-based paradigm generally surpasses its autoregressive counterpart, highlighting the advantage of flexible generation order for protein sequences. We release both AMix-2 and ProteinArena to facilitate open research in protein foundation models.
CUDA Agent: Large-Scale Agentic RL for High-Performance CUDA Kernel Generation
Feb 27, 2026GPU kernel optimization is fundamental to modern deep learning but remains a highly specialized task requiring deep hardware expertise. Despite strong performance in general programming, large language models (LLMs) remain uncompetitive with compiler-based systems such as torch.compile for CUDA kernel generation. Existing CUDA code generation approaches either rely on training-free refinement or fine-tune models within fixed multi-turn execution-feedback loops, but both paradigms fail to fundamentally improve the model's intrinsic CUDA optimization ability, resulting in limited performance gains. We present CUDA Agent, a large-scale agentic reinforcement learning system that develops CUDA kernel expertise through three components: a scalable data synthesis pipeline, a skill-augmented CUDA development environment with automated verification and profiling to provide reliable reward signals, and reinforcement learning algorithmic techniques enabling stable training. CUDA Agent achieves state-of-the-art results on KernelBench, delivering 100\%, 100\%, and 92\% faster rate over torch.compile on KernelBench Level-1, Level-2, and Level-3 splits, outperforming the strongest proprietary models such as Claude Opus 4.5 and Gemini 3 Pro by about 40\% on the hardest Level-3 setting.
S$^2$Drug: Bridging Protein Sequence and 3D Structure in Contrastive Representation Learning for Virtual Screening
Nov 10, 2025Virtual screening (VS) is an essential task in drug discovery, focusing on the identification of small-molecule ligands that bind to specific protein pockets. Existing deep learning methods, from early regression models to recent contrastive learning approaches, primarily rely on structural data while overlooking protein sequences, which are more accessible and can enhance generalizability. However, directly integrating protein sequences poses challenges due to the redundancy and noise in large-scale protein-ligand datasets. To address these limitations, we propose \textbf{S$^2$Drug}, a two-stage framework that explicitly incorporates protein \textbf{S}equence information and 3D \textbf{S}tructure context in protein-ligand contrastive representation learning. In the first stage, we perform protein sequence pretraining on ChemBL using an ESM2-based backbone, combined with a tailored data sampling strategy to reduce redundancy and noise on both protein and ligand sides. In the second stage, we fine-tune on PDBBind by fusing sequence and structure information through a residue-level gating module, while introducing an auxiliary binding site prediction task. This auxiliary task guides the model to accurately localize binding residues within the protein sequence and capture their 3D spatial arrangement, thereby refining protein-ligand matching. Across multiple benchmarks, S$^2$Drug consistently improves virtual screening performance and achieves strong results on binding site prediction, demonstrating the value of bridging sequence and structure in contrastive learning.
FLEX: Continuous Agent Evolution via Forward Learning from Experience
Nov 09, 2025Autonomous agents driven by Large Language Models (LLMs) have revolutionized reasoning and problem-solving but remain static after training, unable to grow with experience as intelligent beings do during deployment. We introduce Forward Learning with EXperience (FLEX), a gradient-free learning paradigm that enables LLM agents to continuously evolve through accumulated experience. Specifically, FLEX cultivates scalable and inheritable evolution by constructing a structured experience library through continual reflection on successes and failures during interaction with the environment. FLEX delivers substantial improvements on mathematical reasoning, chemical retrosynthesis, and protein fitness prediction (up to 23% on AIME25, 10% on USPTO50k, and 14% on ProteinGym). We further identify a clear scaling law of experiential growth and the phenomenon of experience inheritance across agents, marking a step toward scalable and inheritable continuous agent evolution. Project Page: https://flex-gensi-thuair.github.io.
Coder as Editor: Code-driven Interpretable Molecular Optimization
Oct 16, 2025Molecular optimization is a central task in drug discovery that requires precise structural reasoning and domain knowledge. While large language models (LLMs) have shown promise in generating high-level editing intentions in natural language, they often struggle to faithfully execute these modifications-particularly when operating on non-intuitive representations like SMILES. We introduce MECo, a framework that bridges reasoning and execution by translating editing actions into executable code. MECo reformulates molecular optimization for LLMs as a cascaded framework: generating human-interpretable editing intentions from a molecule and property goal, followed by translating those intentions into executable structural edits via code generation. Our approach achieves over 98% accuracy in reproducing held-out realistic edits derived from chemical reactions and target-specific compound pairs. On downstream optimization benchmarks spanning physicochemical properties and target activities, MECo substantially improves consistency by 38-86 percentage points to 90%+ and achieves higher success rates over SMILES-based baselines while preserving structural similarity. By aligning intention with execution, MECo enables consistent, controllable and interpretable molecular design, laying the foundation for high-fidelity feedback loops and collaborative human-AI workflows in drug discovery.
ShortListing Model: A Streamlined SimplexDiffusion for Discrete Variable Generation
Aug 24, 2025Generative modeling of discrete variables is challenging yet crucial for applications in natural language processing and biological sequence design. We introduce the Shortlisting Model (SLM), a novel simplex-based diffusion model inspired by progressive candidate pruning. SLM operates on simplex centroids, reducing generation complexity and enhancing scalability. Additionally, SLM incorporates a flexible implementation of classifier-free guidance, enhancing unconditional generation performance. Extensive experiments on DNA promoter and enhancer design, protein design, character-level and large-vocabulary language modeling demonstrate the competitive performance and strong potential of SLM. Our code can be found at https://github.com/GenSI-THUAIR/SLM
MemAgent: Reshaping Long-Context LLM with Multi-Conv RL-based Memory Agent
Jul 03, 2025

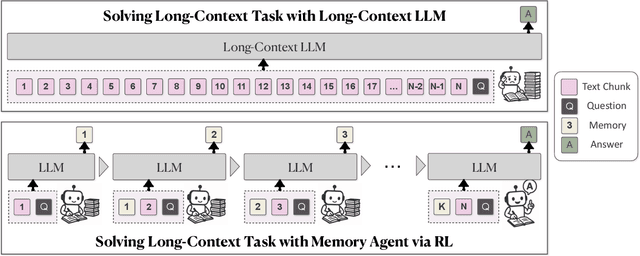
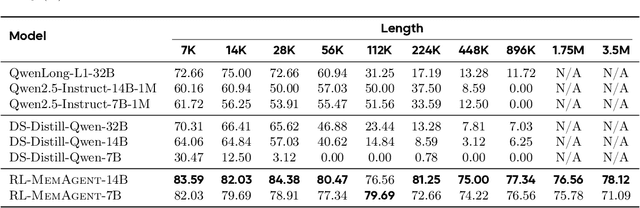
Despite improvements by length extrapolation, efficient attention and memory modules, handling infinitely long documents with linear complexity without performance degradation during extrapolation remains the ultimate challenge in long-text processing. We directly optimize for long-text tasks in an end-to-end fashion and introduce a novel agent workflow, MemAgent, which reads text in segments and updates the memory using an overwrite strategy. We extend the DAPO algorithm to facilitate training via independent-context multi-conversation generation. MemAgent has demonstrated superb long-context capabilities, being able to extrapolate from an 8K context trained on 32K text to a 3.5M QA task with performance loss < 5% and achieves 95%+ in 512K RULER test.
AANet: Virtual Screening under Structural Uncertainty via Alignment and Aggregation
Jun 06, 2025Virtual screening (VS) is a critical component of modern drug discovery, yet most existing methods--whether physics-based or deep learning-based--are developed around holo protein structures with known ligand-bound pockets. Consequently, their performance degrades significantly on apo or predicted structures such as those from AlphaFold2, which are more representative of real-world early-stage drug discovery, where pocket information is often missing. In this paper, we introduce an alignment-and-aggregation framework to enable accurate virtual screening under structural uncertainty. Our method comprises two core components: (1) a tri-modal contrastive learning module that aligns representations of the ligand, the holo pocket, and cavities detected from structures, thereby enhancing robustness to pocket localization error; and (2) a cross-attention based adapter for dynamically aggregating candidate binding sites, enabling the model to learn from activity data even without precise pocket annotations. We evaluated our method on a newly curated benchmark of apo structures, where it significantly outperforms state-of-the-art methods in blind apo setting, improving the early enrichment factor (EF1%) from 11.75 to 37.19. Notably, it also maintains strong performance on holo structures. These results demonstrate the promise of our approach in advancing first-in-class drug discovery, particularly in scenarios lacking experimentally resolved protein-ligand complexes.
Piloting Structure-Based Drug Design via Modality-Specific Optimal Schedule
May 12, 2025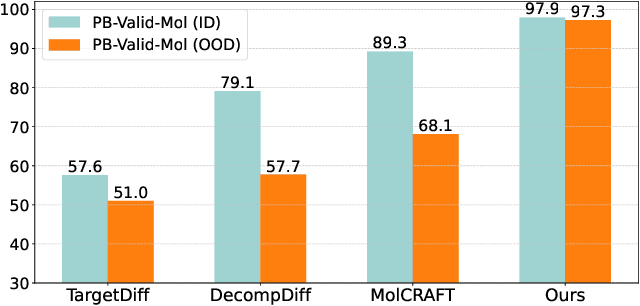
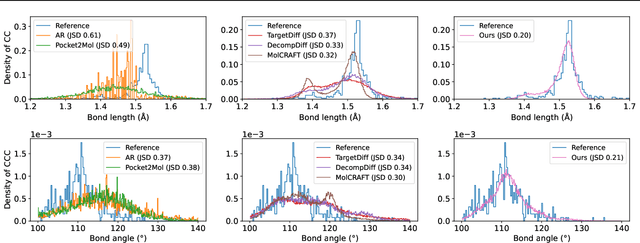
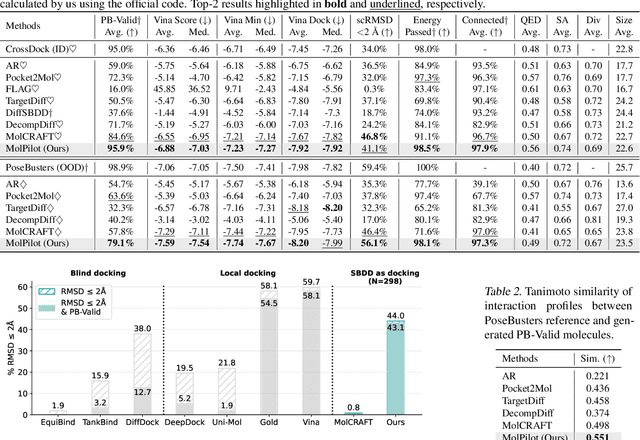
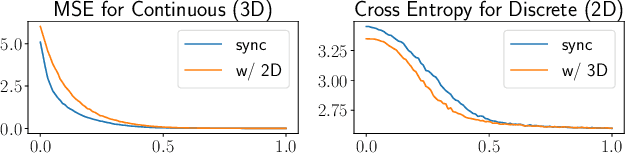
Structure-Based Drug Design (SBDD) is crucial for identifying bioactive molecules. Recent deep generative models are faced with challenges in geometric structure modeling. A major bottleneck lies in the twisted probability path of multi-modalities -- continuous 3D positions and discrete 2D topologies -- which jointly determine molecular geometries. By establishing the fact that noise schedules decide the Variational Lower Bound (VLB) for the twisted probability path, we propose VLB-Optimal Scheduling (VOS) strategy in this under-explored area, which optimizes VLB as a path integral for SBDD. Our model effectively enhances molecular geometries and interaction modeling, achieving state-of-the-art PoseBusters passing rate of 95.9% on CrossDock, more than 10% improvement upon strong baselines, while maintaining high affinities and robust intramolecular validity evaluated on held-out test set.