 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Periodic Bayesian Flow for Material Generation
Feb 04, 2025



Generative modeling of crystal data distribution is an important yet challenging task due to the unique periodic physical symmetry of crystals. Diffusion-based methods have shown early promise in modeling crystal distribution. More recently, Bayesian Flow Networks were introduced to aggregate noisy latent variables, resulting in a variance-reduced parameter space that has been shown to be advantageous for modeling Euclidean data distributions with structural constraints (Song et al., 2023). Inspired by this, we seek to unlock its potential for modeling variables located in non-Euclidean manifolds e.g. those within crystal structures, by overcoming challenging theoretical issues. We introduce CrysBFN, a novel crystal generation method by proposing a periodic Bayesian flow, which essentially differs from the original Gaussian-based BFN by exhibiting non-monotonic entropy dynamics. To successfully realize the concept of periodic Bayesian flow, CrysBFN integrates a new entropy conditioning mechanism and empirically demonstrates its significance compared to time-conditioning. Extensive experiments over both crystal ab initio generation and crystal structure prediction tasks demonstrate the superiority of CrysBFN, which consistently achieves new state-of-the-art on all benchmarks. Surprisingly, we found that CrysBFN enjoys a significant improvement in sampling efficiency, e.g., ~100x speedup 10 v.s. 2000 steps network forwards) compared with previous diffusion-based methods on MP-20 dataset. Code is available at https://github.com/wu-han-lin/CrysBFN.
Structure-Based Molecule Optimization via Gradient-Guided Bayesian Update
Nov 21, 2024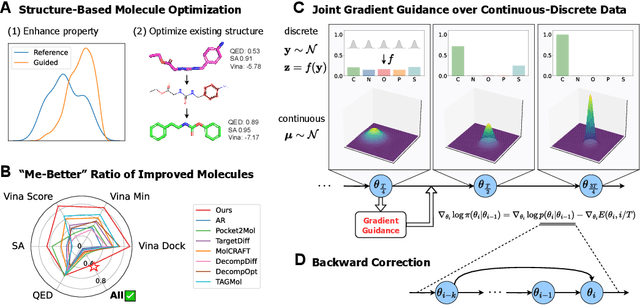
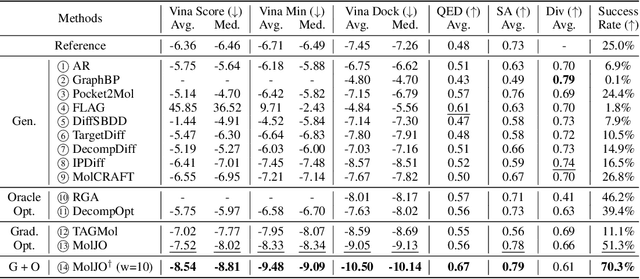
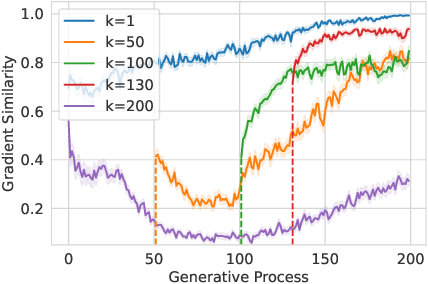
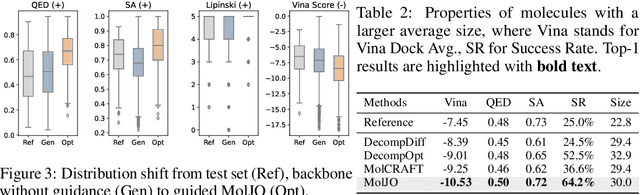
Structure-based molecule optimization (SBMO) aims to optimize molecules with both continuous coordinates and discrete types against protein targets. A promising direction is to exert gradient guidance on generative models given its remarkable success in images, but it is challenging to guide discrete data and risks inconsistencies between modalities. To this end, we leverage a continuous and differentiable space derived through Bayesian inference, presenting Molecule Joint Optimization (MolJO), the first gradient-based SBMO framework that facilitates joint guidance signals across different modalities while preserving SE(3)-equivariance. We introduce a novel backward correction strategy that optimizes within a sliding window of the past histories, allowing for a seamless trade-off between explore-and-exploit during optimization. Our proposed MolJO achieves state-of-the-art performance on CrossDocked2020 benchmark (Success Rate 51.3% , Vina Dock -9.05 and SA 0.78), more than 4x improvement in Success Rate compared to the gradient-based counterpart, and 2x "Me-Better" Ratio as much as 3D baselines. Furthermore, we extend MolJO to a wide range of optimization settings, including multi-objective optimization and challenging tasks in drug design such as R-group optimization and scaffold hopping, further underscoring its versatility and potential.
Equivariant Flow Matching with Hybrid Probability Transport
Dec 12, 2023



The generation of 3D molecules requires simultaneously deciding the categorical features~(atom types) and continuous features~(atom coordinates). Deep generative models, especially Diffusion Models (DMs), have demonstrated effectiveness in generating feature-rich geometries. However, existing DMs typically suffer from unstable probability dynamics with inefficient sampling speed. In this paper, we introduce geometric flow matching, which enjoys the advantages of both equivariant modeling and stabilized probability dynamics. More specifically, we propose a hybrid probability path where the coordinates probability path is regularized by an equivariant optimal transport, and the information between different modalities is aligned. Experimentally, the proposed method could consistently achieve better performance on multiple molecule generation benchmarks with 4.75$\times$ speed up of sampling on average.