 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePAST: A multimodal single-cell foundation model for histopathology and spatial transcriptomics in cancer
Jul 08, 2025While pathology foundation models have transformed cancer image analysis, they often lack integration with molecular data at single-cell resolution, limiting their utility for precision oncology. Here, we present PAST, a pan-cancer single-cell foundation model trained on 20 million paired histopathology images and single-cell transcriptomes spanning multiple tumor types and tissue contexts. By jointly encoding cellular morphology and gene expression, PAST learns unified cross-modal representations that capture both spatial and molecular heterogeneity at the cellular level. This approach enables accurate prediction of single-cell gene expression, virtual molecular staining, and multimodal survival analysis directly from routine pathology slides. Across diverse cancers and downstream tasks, PAST consistently exceeds the performance of existing approaches, demonstrating robust generalizability and scalability. Our work establishes a new paradigm for pathology foundation models, providing a versatile tool for high-resolution spatial omics, mechanistic discovery, and precision cancer research.
ChromFound: Towards A Universal Foundation Model for Single-Cell Chromatin Accessibility Data
May 19, 2025The advent of single-cell Assay for Transposase-Accessible Chromatin using sequencing (scATAC-seq) offers an innovative perspective for deciphering regulatory mechanisms by assembling a vast repository of single-cell chromatin accessibility data. While foundation models have achieved significant success in single-cell transcriptomics, there is currently no foundation model for scATAC-seq that supports zero-shot high-quality cell identification and comprehensive multi-omics analysis simultaneously. Key challenges lie in the high dimensionality and sparsity of scATAC-seq data, as well as the lack of a standardized schema for representing open chromatin regions (OCRs). Here, we present \textbf{ChromFound}, a foundation model tailored for scATAC-seq. ChromFound utilizes a hybrid architecture and genome-aware tokenization to effectively capture genome-wide long contexts and regulatory signals from dynamic chromatin landscapes. Pretrained on 1.97 million cells from 30 tissues and 6 disease conditions, ChromFound demonstrates broad applicability across 6 diverse tasks. Notably, it achieves robust zero-shot performance in generating universal cell representations and exhibits excellent transferability in cell type annotation and cross-omics prediction. By uncovering enhancer-gene links undetected by existing computational methods, ChromFound offers a promising framework for understanding disease risk variants in the noncoding genome.
Structure-Based Molecule Optimization via Gradient-Guided Bayesian Update
Nov 21, 2024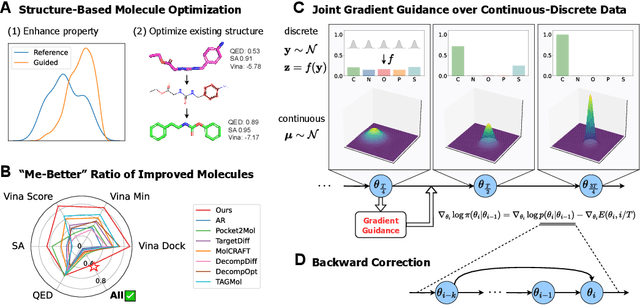
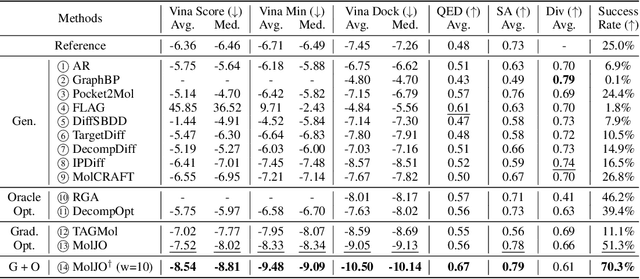
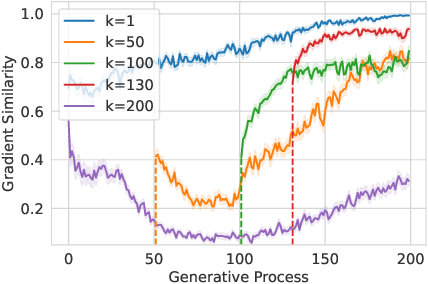
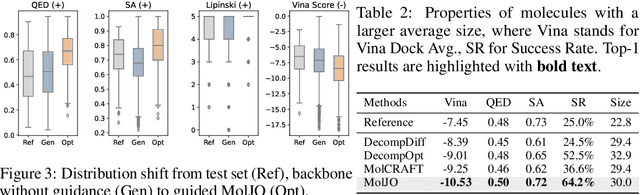
Structure-based molecule optimization (SBMO) aims to optimize molecules with both continuous coordinates and discrete types against protein targets. A promising direction is to exert gradient guidance on generative models given its remarkable success in images, but it is challenging to guide discrete data and risks inconsistencies between modalities. To this end, we leverage a continuous and differentiable space derived through Bayesian inference, presenting Molecule Joint Optimization (MolJO), the first gradient-based SBMO framework that facilitates joint guidance signals across different modalities while preserving SE(3)-equivariance. We introduce a novel backward correction strategy that optimizes within a sliding window of the past histories, allowing for a seamless trade-off between explore-and-exploit during optimization. Our proposed MolJO achieves state-of-the-art performance on CrossDocked2020 benchmark (Success Rate 51.3% , Vina Dock -9.05 and SA 0.78), more than 4x improvement in Success Rate compared to the gradient-based counterpart, and 2x "Me-Better" Ratio as much as 3D baselines. Furthermore, we extend MolJO to a wide range of optimization settings, including multi-objective optimization and challenging tasks in drug design such as R-group optimization and scaffold hopping, further underscoring its versatility and potential.
LangCell: Language-Cell Pre-training for Cell Identity Understanding
May 09, 2024



Cell identity encompasses various semantic aspects of a cell, including cell type, pathway information, disease information, and more, which are essential for biologists to gain insights into its biological characteristics. Understanding cell identity from the transcriptomic data, such as annotating cell types, have become an important task in bioinformatics. As these semantic aspects are determined by human experts, it is impossible for AI models to effectively carry out cell identity understanding tasks without the supervision signals provided by single-cell and label pairs. The single-cell pre-trained language models (PLMs) currently used for this task are trained only on a single modality, transcriptomics data, lack an understanding of cell identity knowledge. As a result, they have to be fine-tuned for downstream tasks and struggle when lacking labeled data with the desired semantic labels. To address this issue, we propose an innovative solution by constructing a unified representation of single-cell data and natural language during the pre-training phase, allowing the model to directly incorporate insights related to cell identity. More specifically, we introduce \textbf{LangCell}, the first \textbf{Lang}uage-\textbf{Cell} pre-training framework. LangCell utilizes texts enriched with cell identity information to gain a profound comprehension of cross-modal knowledge. Results from experiments conducted on different benchmarks show that LangCell is the only single-cell PLM that can work effectively in zero-shot cell identity understanding scenarios, and also significantly outperforms existing models in few-shot and fine-tuning cell identity understanding scenarios.
DeepCRE: Revolutionizing Drug R&D with Cutting-Edge Computational Models
Mar 09, 2024The fields of pharmaceutical development and therapeutic application both face substantial challenges. The therapeutic domain calls for more treatment alternatives, while numerous promising pre-clinical drugs have failed in clinical trials. One of the reasons is the inadequacy of Cross-drug Response Evaluation (CRE) during the late stage of drug development. Although in-silico CRE models offer a solution to this problem, existing methodologies are either limited to early development stages or lack the capacity for a comprehensive CRE analysis. Herein, we introduce a novel computational model named DeepCRE and present the potential of DeepCRE in advancing therapeutic discovery and development. DeepCRE outperforms the existing best models by achieving an average performance improvement of 17.7\% in patient-level CRE and a 5-fold increase in indication-level CRE. Furthermore, DeepCRE has identified six drug candidates that show significantly greater effectiveness than a comparator set of two approved drugs in 5/8 colorectal cancer (CRC) organoids. This highlights DeepCRE's ability to identify a collection of drug candidates with superior therapeutic effects, underscoring its potential to revolutionize the field of therapeutic development.
Empowering AI drug discovery with explicit and implicit knowledge
Apr 17, 2023Motivation: Recently, research on independently utilizing either explicit knowledge from knowledge graphs or implicit knowledge from biomedical literature for AI drug discovery has been growing rapidly. These approaches have greatly improved the prediction accuracy of AI models on multiple downstream tasks. However, integrating explicit and implicit knowledge independently hinders their understanding of molecules. Results: We propose DeepEIK, a unified deep learning framework that incorporates both explicit and implicit knowledge for AI drug discovery. We adopt feature fusion to process the multi-modal inputs, and leverage the attention mechanism to denoise the text information. Experiments show that DeepEIK significantly outperforms state-of-the-art methods on crucial tasks in AI drug discovery including drug-target interaction prediction, drug property prediction and protein-protein interaction prediction. Further studies show that benefiting from explicit and implicit knowledge, our framework achieves a deeper understanding of molecules and shows promising potential in facilitating drug discovery applications.