 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePiloting Structure-Based Drug Design via Modality-Specific Optimal Schedule
May 12, 2025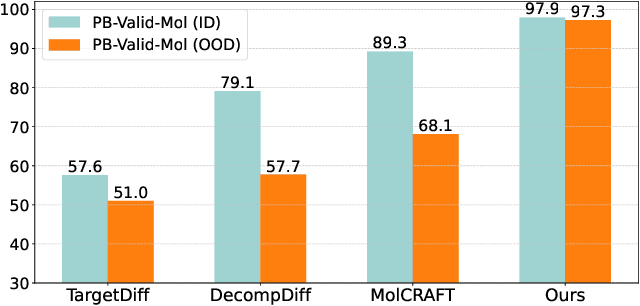
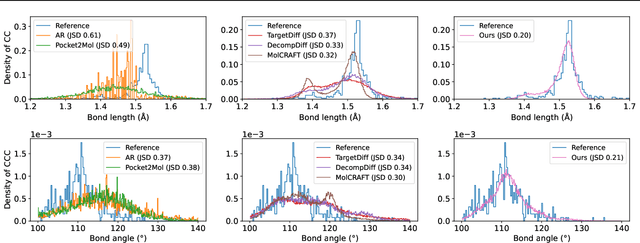
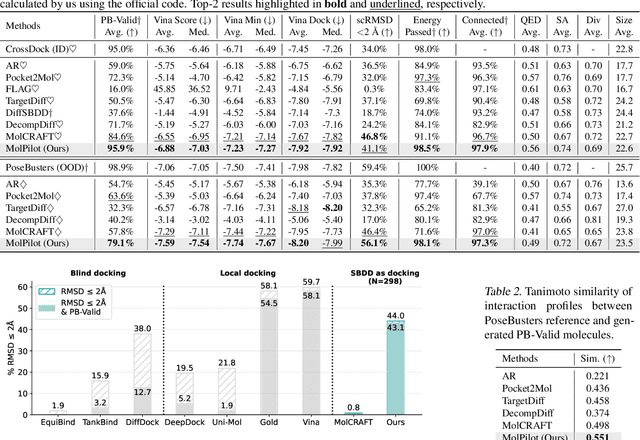
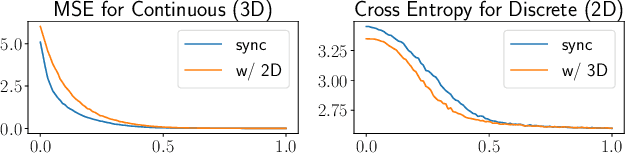
Structure-Based Drug Design (SBDD) is crucial for identifying bioactive molecules. Recent deep generative models are faced with challenges in geometric structure modeling. A major bottleneck lies in the twisted probability path of multi-modalities -- continuous 3D positions and discrete 2D topologies -- which jointly determine molecular geometries. By establishing the fact that noise schedules decide the Variational Lower Bound (VLB) for the twisted probability path, we propose VLB-Optimal Scheduling (VOS) strategy in this under-explored area, which optimizes VLB as a path integral for SBDD. Our model effectively enhances molecular geometries and interaction modeling, achieving state-of-the-art PoseBusters passing rate of 95.9% on CrossDock, more than 10% improvement upon strong baselines, while maintaining high affinities and robust intramolecular validity evaluated on held-out test set.
Structure-Based Molecule Optimization via Gradient-Guided Bayesian Update
Nov 21, 2024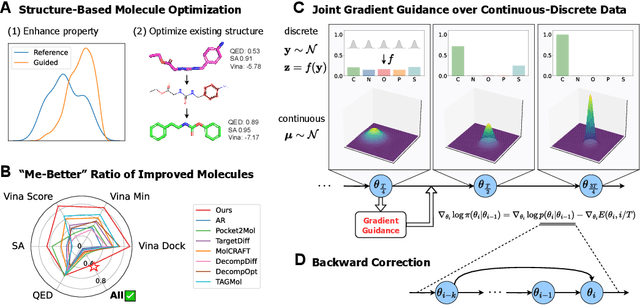
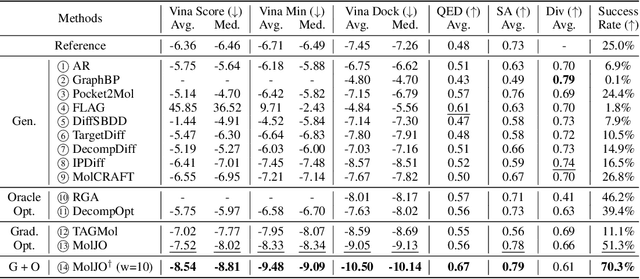
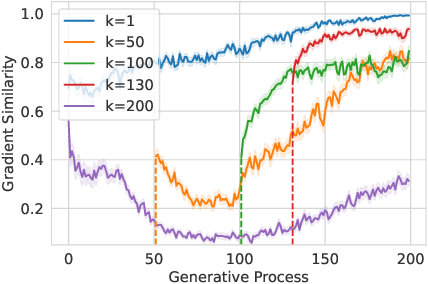
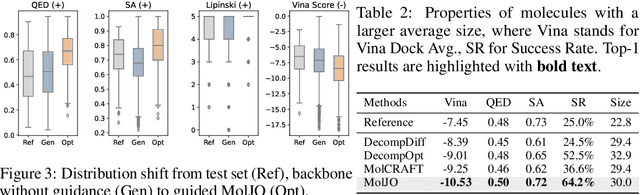
Structure-based molecule optimization (SBMO) aims to optimize molecules with both continuous coordinates and discrete types against protein targets. A promising direction is to exert gradient guidance on generative models given its remarkable success in images, but it is challenging to guide discrete data and risks inconsistencies between modalities. To this end, we leverage a continuous and differentiable space derived through Bayesian inference, presenting Molecule Joint Optimization (MolJO), the first gradient-based SBMO framework that facilitates joint guidance signals across different modalities while preserving SE(3)-equivariance. We introduce a novel backward correction strategy that optimizes within a sliding window of the past histories, allowing for a seamless trade-off between explore-and-exploit during optimization. Our proposed MolJO achieves state-of-the-art performance on CrossDocked2020 benchmark (Success Rate 51.3% , Vina Dock -9.05 and SA 0.78), more than 4x improvement in Success Rate compared to the gradient-based counterpart, and 2x "Me-Better" Ratio as much as 3D baselines. Furthermore, we extend MolJO to a wide range of optimization settings, including multi-objective optimization and challenging tasks in drug design such as R-group optimization and scaffold hopping, further underscoring its versatility and potential.
MolCRAFT: Structure-Based Drug Design in Continuous Parameter Space
Apr 18, 2024Generative models for structure-based drug design (SBDD) have shown promising results in recent years. Existing works mainly focus on how to generate molecules with higher binding affinity, ignoring the feasibility prerequisites for generated 3D poses and resulting in false positives. We conduct thorough studies on key factors of ill-conformational problems when applying autoregressive methods and diffusion to SBDD, including mode collapse and hybrid continuous-discrete space. In this paper, we introduce \ours, the first SBDD model that operates in the continuous parameter space, together with a novel noise reduced sampling strategy. Empirical results show that our model consistently achieves superior performance in binding affinity with more stable 3D structure, demonstrating our ability to accurately model interatomic interactions. To our best knowledge, MolCRAFT is the first to achieve reference-level Vina Scores (-6.59 kcal/mol), outperforming other strong baselines by a wide margin (-0.84 kcal/mol). Code is available at https://github.com/AlgoMole/MolCRAFT.
Manual Evaluation Matters: Reviewing Test Protocols of Distantly Supervised Relation Extraction
May 20, 2021



Distantly supervised (DS) relation extraction (RE) has attracted much attention in the past few years as it can utilize large-scale auto-labeled data. However, its evaluation has long been a problem: previous works either took costly and inconsistent methods to manually examine a small sample of model predictions, or directly test models on auto-labeled data -- which, by our check, produce as much as 53% wrong labels at the entity pair level in the popular NYT10 dataset. This problem has not only led to inaccurate evaluation, but also made it hard to understand where we are and what's left to improve in the research of DS-RE. To evaluate DS-RE models in a more credible way, we build manually-annotated test sets for two DS-RE datasets, NYT10 and Wiki20, and thoroughly evaluate several competitive models, especially the latest pre-trained ones. The experimental results show that the manual evaluation can indicate very different conclusions from automatic ones, especially some unexpected observations, e.g., pre-trained models can achieve dominating performance while being more susceptible to false-positives compared to previous methods. We hope that both our manual test sets and novel observations can help advance future DS-RE research.