 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBatched Contextual Reinforcement: A Task-Scaling Law for Efficient Reasoning
Apr 02, 2026Large Language Models employing Chain-of-Thought reasoning achieve strong performance but suffer from excessive token consumption that inflates inference costs. Existing efficiency methods such as explicit length penalties, difficulty estimators, or multi-stage curricula either degrade reasoning quality or require complex training pipelines. We introduce Batched Contextual Reinforcement, a minimalist, single-stage training paradigm that unlocks efficient reasoning through a simple structural modification: training the model to solve N problems simultaneously within a shared context window, rewarded purely by per-instance accuracy. This formulation creates an implicit token budget that yields several key findings: (1) We identify a novel task-scaling law: as the number of concurrent problems N increases during inference, per-problem token usage decreases monotonically while accuracy degrades far more gracefully than baselines, establishing N as a controllable throughput dimension. (2) BCR challenges the traditional accuracy-efficiency trade-off by demonstrating a "free lunch" phenomenon at standard single-problem inference. Across both 1.5B and 4B model families, BCR reduces token usage by 15.8% to 62.6% while consistently maintaining or improving accuracy across five major mathematical benchmarks. (3) Qualitative analyses reveal emergent self-regulated efficiency, where models autonomously eliminate redundant metacognitive loops without explicit length supervision. (4) Crucially, we empirically demonstrate that implicit budget constraints successfully circumvent the adversarial gradients and catastrophic optimization collapse inherent to explicit length penalties, offering a highly stable, constraint-based alternative for length control. These results prove BCR practical, showing simple structural incentives unlock latent high-density reasoning in LLMs.
Structure-Based Molecule Optimization via Gradient-Guided Bayesian Update
Nov 21, 2024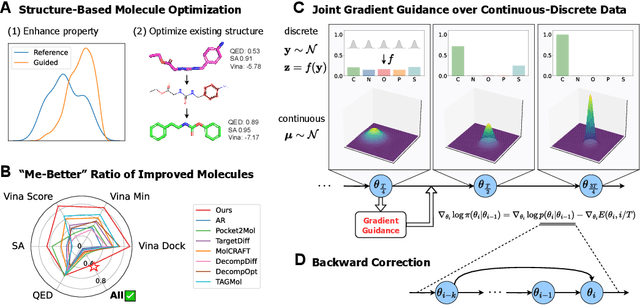
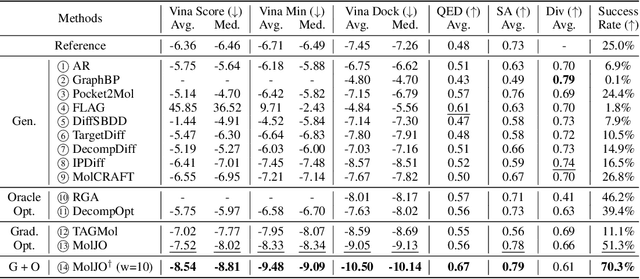
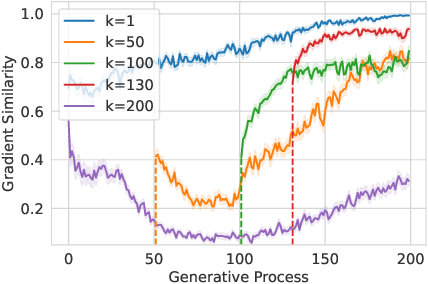
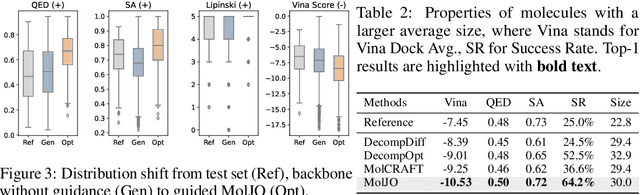
Structure-based molecule optimization (SBMO) aims to optimize molecules with both continuous coordinates and discrete types against protein targets. A promising direction is to exert gradient guidance on generative models given its remarkable success in images, but it is challenging to guide discrete data and risks inconsistencies between modalities. To this end, we leverage a continuous and differentiable space derived through Bayesian inference, presenting Molecule Joint Optimization (MolJO), the first gradient-based SBMO framework that facilitates joint guidance signals across different modalities while preserving SE(3)-equivariance. We introduce a novel backward correction strategy that optimizes within a sliding window of the past histories, allowing for a seamless trade-off between explore-and-exploit during optimization. Our proposed MolJO achieves state-of-the-art performance on CrossDocked2020 benchmark (Success Rate 51.3% , Vina Dock -9.05 and SA 0.78), more than 4x improvement in Success Rate compared to the gradient-based counterpart, and 2x "Me-Better" Ratio as much as 3D baselines. Furthermore, we extend MolJO to a wide range of optimization settings, including multi-objective optimization and challenging tasks in drug design such as R-group optimization and scaffold hopping, further underscoring its versatility and potential.
SIU: A Million-Scale Structural Small Molecule-Protein Interaction Dataset for Unbiased Bioactivity Prediction
Jun 13, 2024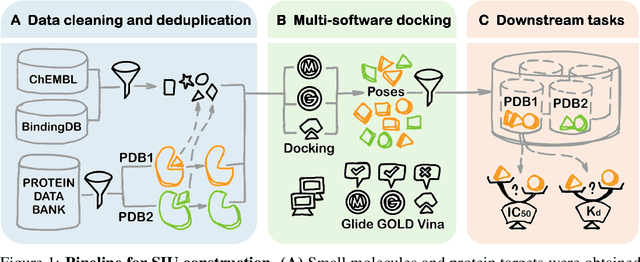

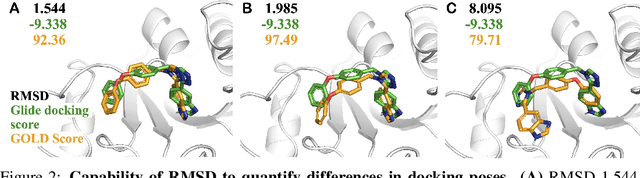
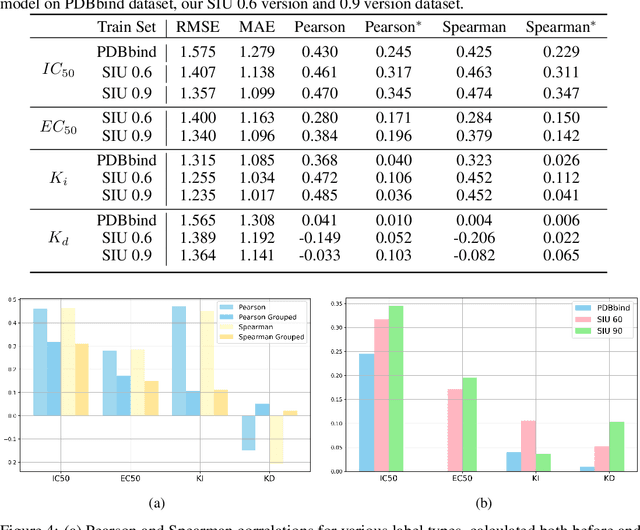
Small molecules play a pivotal role in modern medicine, and scrutinizing their interactions with protein targets is essential for the discovery and development of novel, life-saving therapeutics. The term "bioactivity" encompasses various biological effects resulting from these interactions, including both binding and functional responses. The magnitude of bioactivity dictates the therapeutic or toxic pharmacological outcomes of small molecules, rendering accurate bioactivity prediction crucial for the development of safe and effective drugs. However, existing structural datasets of small molecule-protein interactions are often limited in scale and lack systematically organized bioactivity labels, thereby impeding our understanding of these interactions and precise bioactivity prediction. In this study, we introduce a comprehensive dataset of small molecule-protein interactions, consisting of over a million binding structures, each annotated with real biological activity labels. This dataset is designed to facilitate unbiased bioactivity prediction. We evaluated several classical models on this dataset, and the results demonstrate that the task of unbiased bioactivity prediction is challenging yet essential.