 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning-based density-equalizing map
Jun 10, 2025Density-equalizing map (DEM) serves as a powerful technique for creating shape deformations with the area changes reflecting an underlying density function. In recent decades, DEM has found widespread applications in fields such as data visualization, geometry processing, and medical imaging. Traditional approaches to DEM primarily rely on iterative numerical solvers for diffusion equations or optimization-based methods that minimize handcrafted energy functionals. However, these conventional techniques often face several challenges: they may suffer from limited accuracy, produce overlapping artifacts in extreme cases, and require substantial algorithmic redesign when extended from 2D to 3D, due to the derivative-dependent nature of their energy formulations. In this work, we propose a novel learning-based density-equalizing mapping framework (LDEM) using deep neural networks. Specifically, we introduce a loss function that enforces density uniformity and geometric regularity, and utilize a hierarchical approach to predict the transformations at both the coarse and dense levels. Our method demonstrates superior density-equalizing and bijectivity properties compared to prior methods for a wide range of simple and complex density distributions, and can be easily applied to surface remeshing with different effects. Also, it generalizes seamlessly from 2D to 3D domains without structural changes to the model architecture or loss formulation. Altogether, our work opens up new possibilities for scalable and robust computation of density-equalizing maps for practical applications.
Sentinel: Attention Probing of Proxy Models for LLM Context Compression with an Understanding Perspective
May 29, 2025Retrieval-augmented generation (RAG) enhances large language models (LLMs) with external context, but retrieved passages are often lengthy, noisy, or exceed input limits. Existing compression methods typically require supervised training of dedicated compression models, increasing cost and reducing portability. We propose Sentinel, a lightweight sentence-level compression framework that reframes context filtering as an attention-based understanding task. Rather than training a compression model, Sentinel probes decoder attention from an off-the-shelf 0.5B proxy LLM using a lightweight classifier to identify sentence relevance. Empirically, we find that query-context relevance estimation is consistent across model scales, with 0.5B proxies closely matching the behaviors of larger models. On the LongBench benchmark, Sentinel achieves up to 5$\times$ compression while matching the QA performance of 7B-scale compression systems. Our results suggest that probing native attention signals enables fast, effective, and question-aware context compression. Code available at: https://github.com/yzhangchuck/Sentinel.
PharmAgents: Building a Virtual Pharma with Large Language Model Agents
Mar 31, 2025The discovery of novel small molecule drugs remains a critical scientific challenge with far-reaching implications for treating diseases and advancing human health. Traditional drug development--especially for small molecule therapeutics--is a highly complex, resource-intensive, and time-consuming process that requires multidisciplinary collaboration. Recent breakthroughs in artificial intelligence (AI), particularly the rise of large language models (LLMs), present a transformative opportunity to streamline and accelerate this process. In this paper, we introduce PharmAgents, a virtual pharmaceutical ecosystem driven by LLM-based multi-agent collaboration. PharmAgents simulates the full drug discovery workflow--from target discovery to preclinical evaluation--by integrating explainable, LLM-driven agents equipped with specialized machine learning models and computational tools. Through structured knowledge exchange and automated optimization, PharmAgents identifies potential therapeutic targets, discovers promising lead compounds, enhances binding affinity and key molecular properties, and performs in silico analyses of toxicity and synthetic feasibility. Additionally, the system supports interpretability, agent interaction, and self-evolvement, enabling it to refine future drug designs based on prior experience. By showcasing the potential of LLM-powered multi-agent systems in drug discovery, this work establishes a new paradigm for autonomous, explainable, and scalable pharmaceutical research, with future extensions toward comprehensive drug lifecycle management.
Large Language Models Are Universal Recommendation Learners
Feb 05, 2025



In real-world recommender systems, different tasks are typically addressed using supervised learning on task-specific datasets with carefully designed model architectures. We demonstrate that large language models (LLMs) can function as universal recommendation learners, capable of handling multiple tasks within a unified input-output framework, eliminating the need for specialized model designs. To improve the recommendation performance of LLMs, we introduce a multimodal fusion module for item representation and a sequence-in-set-out approach for efficient candidate generation. When applied to industrial-scale data, our LLM achieves competitive results with expert models elaborately designed for different recommendation tasks. Furthermore, our analysis reveals that recommendation outcomes are highly sensitive to text input, highlighting the potential of prompt engineering in optimizing industrial-scale recommender systems.
Dynamic Attention-Guided Context Decoding for Mitigating Context Faithfulness Hallucinations in Large Language Models
Jan 02, 2025



Large language models (LLMs) often suffer from context faithfulness hallucinations, where outputs deviate from retrieved information due to insufficient context utilization and high output uncertainty. Our uncertainty evaluation experiments reveal a strong correlation between high uncertainty and hallucinations. We hypothesize that attention mechanisms encode signals indicative of contextual utilization, validated through probing analysis. Based on these insights, we propose Dynamic Attention-Guided Context Decoding (DAGCD), a lightweight framework that integrates attention distributions and uncertainty signals in a single-pass decoding process. Experiments across QA datasets demonstrate DAGCD's effectiveness, achieving significant improvements in faithfulness and robustness while maintaining computational efficiency.
From Theory to Therapy: Reframing SBDD Model Evaluation via Practical Metrics
Jun 13, 2024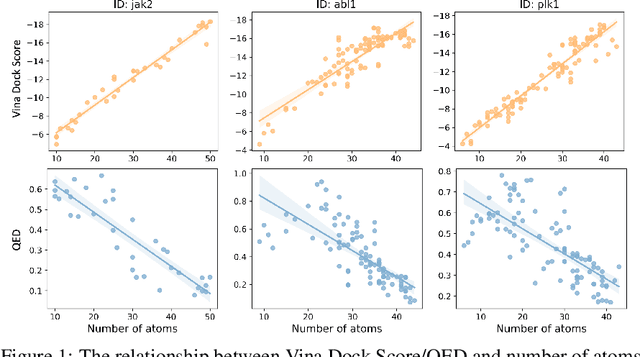
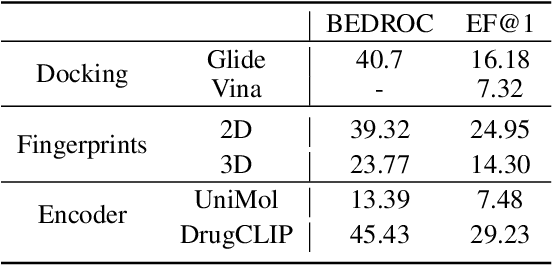
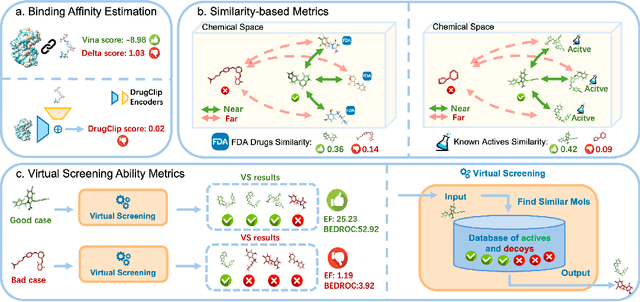
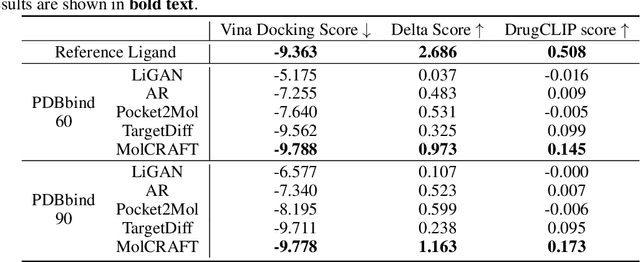
Recent advancements in structure-based drug design (SBDD) have significantly enhanced the efficiency and precision of drug discovery by generating molecules tailored to bind specific protein pockets. Despite these technological strides, their practical application in real-world drug development remains challenging due to the complexities of synthesizing and testing these molecules. The reliability of the Vina docking score, the current standard for assessing binding abilities, is increasingly questioned due to its susceptibility to overfitting. To address these limitations, we propose a comprehensive evaluation framework that includes assessing the similarity of generated molecules to known active compounds, introducing a virtual screening-based metric for practical deployment capabilities, and re-evaluating binding affinity more rigorously. Our experiments reveal that while current SBDD models achieve high Vina scores, they fall short in practical usability metrics, highlighting a significant gap between theoretical predictions and real-world applicability. Our proposed metrics and dataset aim to bridge this gap, enhancing the practical applicability of future SBDD models and aligning them more closely with the needs of pharmaceutical research and development.
SIU: A Million-Scale Structural Small Molecule-Protein Interaction Dataset for Unbiased Bioactivity Prediction
Jun 13, 2024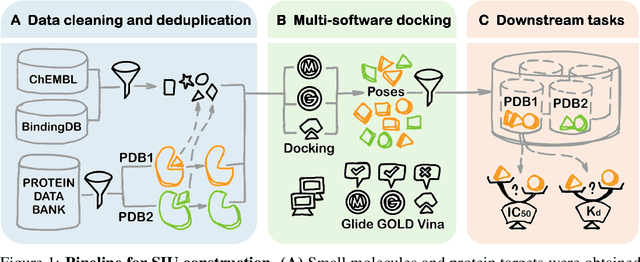

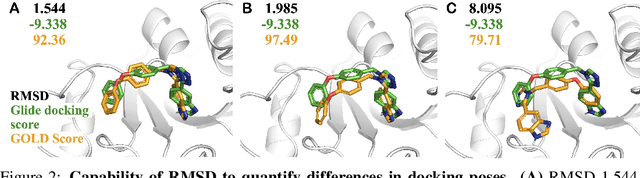
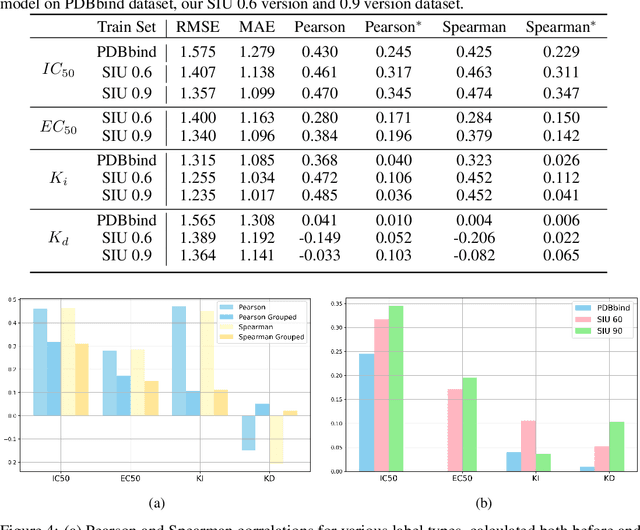
Small molecules play a pivotal role in modern medicine, and scrutinizing their interactions with protein targets is essential for the discovery and development of novel, life-saving therapeutics. The term "bioactivity" encompasses various biological effects resulting from these interactions, including both binding and functional responses. The magnitude of bioactivity dictates the therapeutic or toxic pharmacological outcomes of small molecules, rendering accurate bioactivity prediction crucial for the development of safe and effective drugs. However, existing structural datasets of small molecule-protein interactions are often limited in scale and lack systematically organized bioactivity labels, thereby impeding our understanding of these interactions and precise bioactivity prediction. In this study, we introduce a comprehensive dataset of small molecule-protein interactions, consisting of over a million binding structures, each annotated with real biological activity labels. This dataset is designed to facilitate unbiased bioactivity prediction. We evaluated several classical models on this dataset, and the results demonstrate that the task of unbiased bioactivity prediction is challenging yet essential.
UniCorn: A Unified Contrastive Learning Approach for Multi-view Molecular Representation Learning
May 15, 2024



Recently, a noticeable trend has emerged in developing pre-trained foundation models in the domains of CV and NLP. However, for molecular pre-training, there lacks a universal model capable of effectively applying to various categories of molecular tasks, since existing prevalent pre-training methods exhibit effectiveness for specific types of downstream tasks. Furthermore, the lack of profound understanding of existing pre-training methods, including 2D graph masking, 2D-3D contrastive learning, and 3D denoising, hampers the advancement of molecular foundation models. In this work, we provide a unified comprehension of existing pre-training methods through the lens of contrastive learning. Thus their distinctions lie in clustering different views of molecules, which is shown beneficial to specific downstream tasks. To achieve a complete and general-purpose molecular representation, we propose a novel pre-training framework, named UniCorn, that inherits the merits of the three methods, depicting molecular views in three different levels. SOTA performance across quantum, physicochemical, and biological tasks, along with comprehensive ablation study, validate the universality and effectiveness of UniCorn.