 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePiloting Structure-Based Drug Design via Modality-Specific Optimal Schedule
May 12, 2025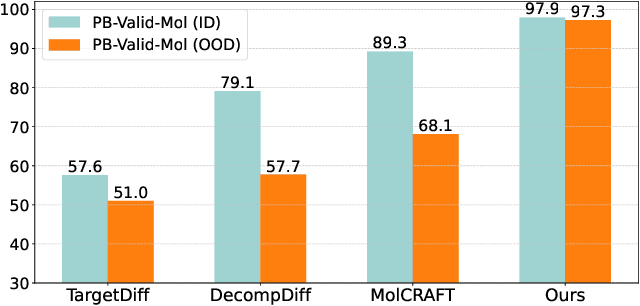
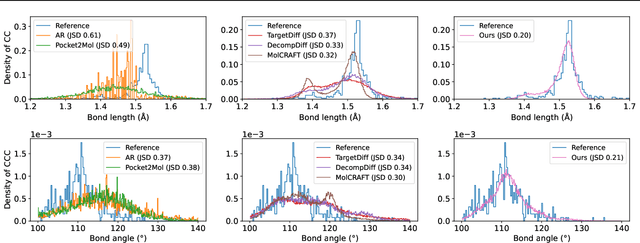
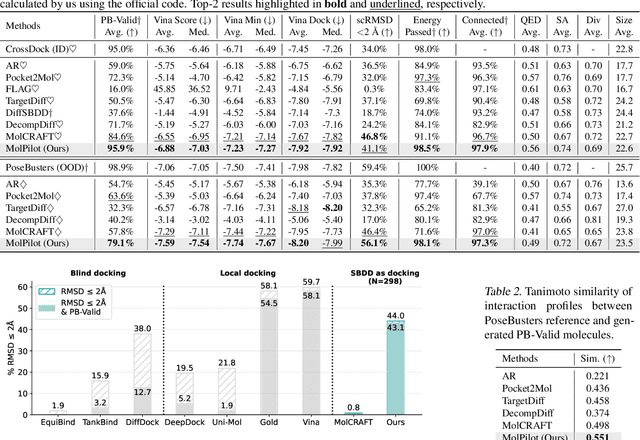
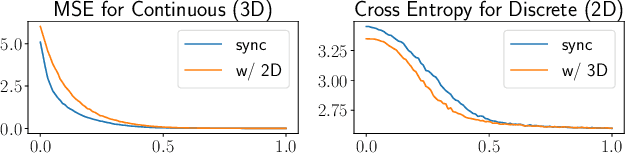
Structure-Based Drug Design (SBDD) is crucial for identifying bioactive molecules. Recent deep generative models are faced with challenges in geometric structure modeling. A major bottleneck lies in the twisted probability path of multi-modalities -- continuous 3D positions and discrete 2D topologies -- which jointly determine molecular geometries. By establishing the fact that noise schedules decide the Variational Lower Bound (VLB) for the twisted probability path, we propose VLB-Optimal Scheduling (VOS) strategy in this under-explored area, which optimizes VLB as a path integral for SBDD. Our model effectively enhances molecular geometries and interaction modeling, achieving state-of-the-art PoseBusters passing rate of 95.9% on CrossDock, more than 10% improvement upon strong baselines, while maintaining high affinities and robust intramolecular validity evaluated on held-out test set.
P2DFlow: A Protein Ensemble Generative Model with SE(3) Flow Matching
Nov 26, 2024Biological processes, functions, and properties are intricately linked to the ensemble of protein conformations, rather than being solely determined by a single stable conformation. In this study, we have developed P2DFlow, a generative model based on SE(3) flow matching, to predict the structural ensembles of proteins. We specifically designed a valuable prior for the flow process and enhanced the model's ability to distinguish each intermediate state by incorporating an additional dimension to describe the ensemble data, which can reflect the physical laws governing the distribution of ensembles, so that the prior knowledge can effectively guide the generation process. When trained and evaluated on the MD datasets of ATLAS, P2DFlow outperforms other baseline models on extensive experiments, successfully capturing the observable dynamic fluctuations as evidenced in crystal structure and MD simulations. As a potential proxy agent for protein molecular simulation, the high-quality ensembles generated by P2DFlow could significantly aid in understanding protein functions across various scenarios. Code is available at https://github.com/BLEACH366/P2DFlow.
Structure-Based Molecule Optimization via Gradient-Guided Bayesian Update
Nov 21, 2024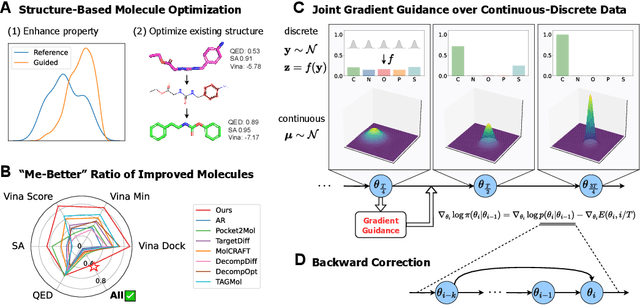
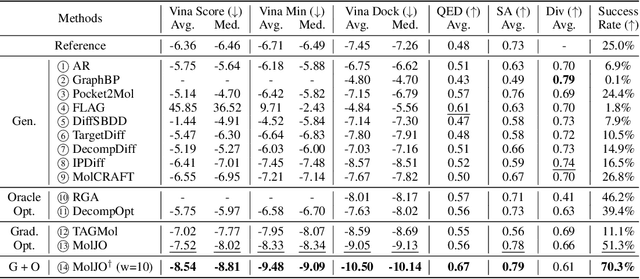
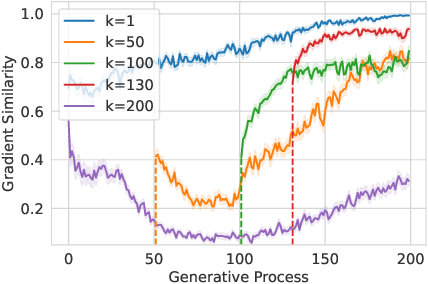
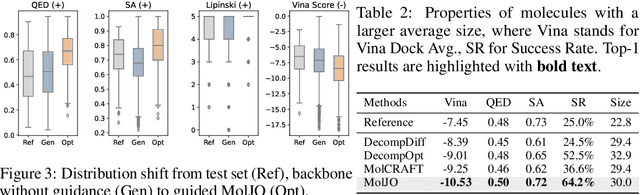
Structure-based molecule optimization (SBMO) aims to optimize molecules with both continuous coordinates and discrete types against protein targets. A promising direction is to exert gradient guidance on generative models given its remarkable success in images, but it is challenging to guide discrete data and risks inconsistencies between modalities. To this end, we leverage a continuous and differentiable space derived through Bayesian inference, presenting Molecule Joint Optimization (MolJO), the first gradient-based SBMO framework that facilitates joint guidance signals across different modalities while preserving SE(3)-equivariance. We introduce a novel backward correction strategy that optimizes within a sliding window of the past histories, allowing for a seamless trade-off between explore-and-exploit during optimization. Our proposed MolJO achieves state-of-the-art performance on CrossDocked2020 benchmark (Success Rate 51.3% , Vina Dock -9.05 and SA 0.78), more than 4x improvement in Success Rate compared to the gradient-based counterpart, and 2x "Me-Better" Ratio as much as 3D baselines. Furthermore, we extend MolJO to a wide range of optimization settings, including multi-objective optimization and challenging tasks in drug design such as R-group optimization and scaffold hopping, further underscoring its versatility and potential.
MolCRAFT: Structure-Based Drug Design in Continuous Parameter Space
Apr 18, 2024Generative models for structure-based drug design (SBDD) have shown promising results in recent years. Existing works mainly focus on how to generate molecules with higher binding affinity, ignoring the feasibility prerequisites for generated 3D poses and resulting in false positives. We conduct thorough studies on key factors of ill-conformational problems when applying autoregressive methods and diffusion to SBDD, including mode collapse and hybrid continuous-discrete space. In this paper, we introduce \ours, the first SBDD model that operates in the continuous parameter space, together with a novel noise reduced sampling strategy. Empirical results show that our model consistently achieves superior performance in binding affinity with more stable 3D structure, demonstrating our ability to accurately model interatomic interactions. To our best knowledge, MolCRAFT is the first to achieve reference-level Vina Scores (-6.59 kcal/mol), outperforming other strong baselines by a wide margin (-0.84 kcal/mol). Code is available at https://github.com/AlgoMole/MolCRAFT.
Unified Generative Modeling of 3D Molecules via Bayesian Flow Networks
Mar 17, 2024Advanced generative model (e.g., diffusion model) derived from simplified continuity assumptions of data distribution, though showing promising progress, has been difficult to apply directly to geometry generation applications due to the multi-modality and noise-sensitive nature of molecule geometry. This work introduces Geometric Bayesian Flow Networks (GeoBFN), which naturally fits molecule geometry by modeling diverse modalities in the differentiable parameter space of distributions. GeoBFN maintains the SE-(3) invariant density modeling property by incorporating equivariant inter-dependency modeling on parameters of distributions and unifying the probabilistic modeling of different modalities. Through optimized training and sampling techniques, we demonstrate that GeoBFN achieves state-of-the-art performance on multiple 3D molecule generation benchmarks in terms of generation quality (90.87% molecule stability in QM9 and 85.6% atom stability in GEOM-DRUG. GeoBFN can also conduct sampling with any number of steps to reach an optimal trade-off between efficiency and quality (e.g., 20-times speedup without sacrificing performance).
Structure-Based Drug Design via 3D Molecular Generative Pre-training and Sampling
Feb 22, 2024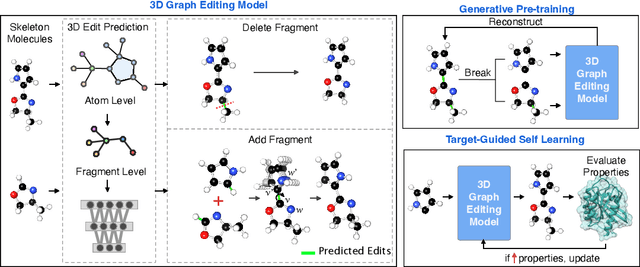
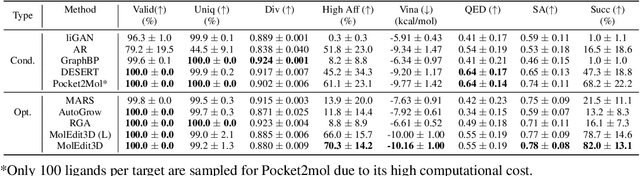
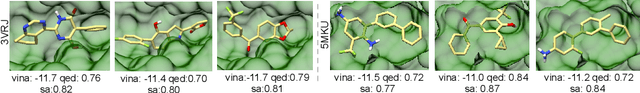
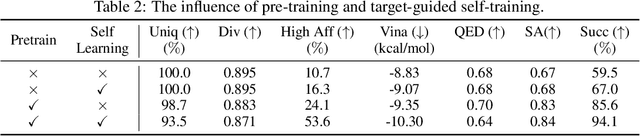
Structure-based drug design aims at generating high affinity ligands with prior knowledge of 3D target structures. Existing methods either use conditional generative model to learn the distribution of 3D ligands given target binding sites, or iteratively modify molecules to optimize a structure-based activity estimator. The former is highly constrained by data quantity and quality, which leaves optimization-based approaches more promising in practical scenario. However, existing optimization-based approaches choose to edit molecules in 2D space, and use molecular docking to estimate the activity using docking predicted 3D target-ligand complexes. The misalignment between the action space and the objective hinders the performance of these models, especially for those employ deep learning for acceleration. In this work, we propose MolEdit3D to combine 3D molecular generation with optimization frameworks. We develop a novel 3D graph editing model to generate molecules using fragments, and pre-train this model on abundant 3D ligands for learning target-independent properties. Then we employ a target-guided self-learning strategy to improve target-related properties using self-sampled molecules. MolEdit3D achieves state-of-the-art performance on majority of the evaluation metrics, and demonstrate strong capability of capturing both target-dependent and -independent properties.